
Abstract
Plasma activated water has shown great promise in a number of emerging application domains; yet the interaction between non-equilibrium plasma and liquid represents a complex multiphase process that is difficult to probe experimentally, necessitating the development of accurate numerical models. In this work, a global computational model was developed to follow the concentrations of aqueous reactive species in water treated using a surface barrier discharge in ambient air. While the two-film theory has long superseded other methods of modelling mass transfer in such areas of research as environmental and aerosol science, plasma modelling studies continue to use equilibrium and one-film theories. The transport of reactive species across the gas–liquid interface was therefore treated using the one-film and two-film theories, with the results compared to ascertain which is most appropriate for PAW modelling studies. Comparing the model-predicted concentrations to those measured, it was shown that concentrations of aqueous H+ and NO3− ions were better represented by the two-film theory, more closely fitting experimental measurements in trend and in magnitude by a factor of ten, while HNO2 and NO2− showed a slightly worse fit using this theory. This is attributed to the assumption in two-film theory of a gas-phase stagnant film which provides additional resistance to the absorption of hydrophilic species, which is absent in the one-film theory, which could be improved with a more accurate value of the Sherwood number for each species.
Similar content being viewed by others
Introduction
In recent years, Cold Atmospheric Plasmas (CAPs) have been used to treat water, generating a chemically active solution that became known as Plasma Activated Water (PAW) [1]. The chemical activity of PAW makes it suitable for a diverse range of applications, including the stimulation of crop growth [2, 3], food processing [4], bacterial inactivation [5], and waste-water treatment [6]. To create PAW, plasma is typically generated at atmospheric pressure using various types of discharge, such as surface barrier discharge (SBD) or a plasma jet [7]. The chemical species generated in the plasma can then either diffuse, be carried by convection, or be generated locally at the gas–liquid interface, where they dissolve into the water. Subsequently, they react chemically with the water, generating a wide range of reactive species in the aqueous phase [8].
A common aspect in all the PAW applications mentioned previously is the need to control the dose of reactive species dissolving into water, which requires accurate determination of their quantities. Various techniques with different selectivity and sensitivity have been used for the investigation and quantification of long lifetime species in the liquid phase such as NO2−, NO3− and H2O2. The most common experimental technique is the use of spectrophotometric assays, which are based on the affinity between a chemical reagent and one of the species under analysis. The intensity of the coloured complex can then be compared to a standard curve. Other techniques reported include fluorescence—in which chemical compounds are mixed with fluorescein dyes—as well as ion and liquid chromatography [7]. However, each diagnostic technique is typically only applicable to quantify a limited number of species and thus cannot provide information on all species present in the aqueous phase. In addition, diagnostic techniques provide no information on the chemical pathways which result in the formation of species; this makes it difficult to unravel the link between the plasma parameters and the final composition of the created PAW. Consequently, experimentally validated numerical modelling is needed to obtain further information on the concentrations of generated species and the chemical pathways involved.
Many experiments have treatment times of the order of tens of minutes [1,2,3,4]. For numerical modelling to be performed on a similar timescale, it is necessary that the model be as simple as possible without sacrificing accuracy. To capture the numerous processes that take place in mass transfer from a gaseous phase to a liquid phase—such as diffusion, surface adhesion, eddy penetration, etc.—many models include one or two dimensions in space, giving a more accurate description of the modelled configuration. However, including even one spatial dimension greatly increases the number of degrees of freedom to be solved for, and thus the computing power and running time needed to reach a solution, a problem which worsens significantly as the number of dimensions increase. While a 0D computational model of a typical laboratory PAW system usually takes hours to simulate a few seconds of an experiment, this increases to days and even weeks if a 1D or 2D model is used instead [9].
Fast yet accurate alternative modelling methods are highly desirable. Global models (also referred to as 0D models) run on spatially averaged domains, which significantly simplifies the equations to be solved, thus leading to low computational cost. This advantage makes them ideal for long timescale modelling of PAW. By their very nature, such 0D models are simplifications as they are unable to capture spatially-dependent phenomena and, if used without considering the impact of such a simplification on the results, can produce poor agreement with experimental results. This problem can be mitigated by combining the 0D model with theories that describe the overall effects of the spatiality dependent phenomena without computing them explicitly, which would require consideration of at least one spatial dimension.
In the context of PAW modelling, one shortcoming of 0D models is the difficulty of accounting for rapidly reacting species which react on timescales shorter than the transport timescales in the water. While this limits the accuracy of 0D models, it is less important in circumstances where only long-lived species from the gas phase arrive at the surface, such as discharge configurations where the distance between the plasma and the liquid is large enough. In such cases, the only mechanism behind the presence of the rapidly reacting species in the liquid is local generation through chemical reactions.
A potential mitigation for this shortcoming is separating the bulk liquid into a number of discrete layers, with a flux calculated between each layer [10]. Such an approach will increase the complexity of the model, but to a very small extent when compared with the complexity of a 1D model.
A further problem is describing the flux of plasma-generated species across the gas–liquid interface. Theories describing the uptake of gaseous species into the liquid can be grouped based on the simplicity of description into three broad categories: zero-film, one-film, and two-film interfacial transport theories [11]. Each of these theories has a varying extent to which it will account for the several physical processes that contribute to the absorption of species, summarising them into a single equation for each species, greatly simplifying the calculation of flux from species densities across the interface.
The simplest descriptions of gas transport across an interface are provided by zero-film theories, in which there is no resistance to the flux of gaseous species into the liquid and the concentration of each aqueous species is in an instantaneous equilibrium with its gaseous counterpart. Such a scenario is described by Henry’s law [12]. Considering that the densities of the plasma generated species change on timescales ranging from nanoseconds to minutes [13], the assumption of instantaneous equilibrium at the surface is unjustified. Plasma species incident on the water surface may not be immediately absorbed into the bulk liquid. Several intermediate processes take place, such as adsorption, whereby the species adhere to the water surface and may either diffuse into the liquid bulk or detach back into the gas. The adsorbed species may also penetrate the surface in bulk packets of eddy currents rather than diffusing gradually [14]. Since the absorption of plasma species into the liquid is not instantaneous, it is not reasonable to assume that equilibrium is achieved instantaneously. Therefore, such a treatment is at best inaccurate when combined with 0D models to describe PAW.
A more general treatment of the gas transport across a gas–liquid interface is provided by one-film theory, which assumes the existence of a “stagnant film” of liquid at the interface in which species are absorbed before diffusing into the liquid bulk. The presence of the stagnant film causes resistance to the flux of the species across the interface as their concentrations increase in the film until equilibrium is reached with the gas phase, preventing any further absorption until the concentration in the stagnant film is reduced again by diffusion [11].
A further upgrade is found in the two-film theory, which posits that two stagnant films exist at the interface: one on the gas side and another on the liquid side [15]. The presence of the stagnant film on the gas side limits the transfer of hydrophilic species—which would have reached the interface and instantly dissolved according to the one-film theories—leading to an accumulation of species in the gaseous stagnant film slowing their rate of uptake into the liquid phase. Similarly, hydrophobic species generated in the liquid phase experience slowing of desorption from the liquid phase [11].
The two-film theory has been the preferred method of modelling gas exchange at the water surface for many years in other areas of scientific research, such as environmental [16,17,18] and aerosol chemistry [19]. A review of publications reveals that this is not the case in the field of PAW, where researchers typically rely on the implementation of spatial dimensions into at least one region of the plasma–air–water system with an equilibrium law at the water surface to model the uptake of plasma induced species.
Recent works on the subject of modelling include those of Liu et al., who developed a model for predicting the aqueous concentrations of species during indirect exposure to an SBD air plasma source over a time of 100 s [20], later following up on this work to predict how the concentrations of species in the PAW created would change over the course of up to 350 h after plasma treatment [21]. Lietz et al. similarly modelled SBD in air but with plasma discharge taking place directly onto the water surface, treating the water as the grounded electrode, to determine the evolution of the aqueous species concentrations during and after a period of 5000 plasma pulses lasting 10 s [22]. This model was later applied to the generation of PAW from an aerosol of water droplets in an air plasma discharge [23]. The use of film theories in modelling the absorption of gases into still water and aerosols are highly applicable to each of these situations.
Verlackt et al. investigated the use of an argon plasma jet to generate PAW, crafting a computational model to ascertain the species concentrations over an exposure time of 60 s [24]. Their model used a 2D axisymmetric fluid approach to describing the plasma jet incident on the water surface, with transport of plasma species into the liquid governed by Henry’s law. They later adapted the model so that, while the fluid flow was still computed in two dimensions, the chemistry in each phase was modelled using a spatially-averaged global model in order to dramatically reduce the impractically long running time [9], emphasising the need for development of such simplified models in the PAW community.
In this study, the influence of the theory chosen to describe the interfacial fluxes of the species on the concentration of aqueous species is explored. This is done using a 0D model describing plasma activation of water using a surface barrier discharge (SBD). To quantify the influence of the adopted interfacial theory on the model, its predictions were compared to experimental measurements of the concentrations of various aqueous species in a PAW solution created under conditions matching those modelled.
Experimental Setup
Plasma Reactor
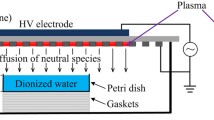
The experimental setup used to validate the models is as shown in Fig. 1. A petri dish of interior diameter 75 mm was filled with 20 ml of deionised water with a conductivity of 2 ± 0.5 µS m−1, leading to a water depth of dl = 4.5 mm and surface area of Aw = 4 420 mm2. A magnetic stir bar was immersed in the water, allowing the water to be well-stirred throughout the experiment. Critically, the stirring aims to make the distribution of reactive species more uniform in the bulk liquid, which is a key assumption in any 0D model.

a Cross section of the experiment setup. Long-lived species generated by the discharge diffuse across the air gap and dissolve in the water. b Photograph of the air plasma generated in the mesh
The SBD reactor was contained in a polymeric housing which included a sealing ring matching the dimensions of the petri dish rim. This gave a snug fit when placed on top of the petri dish ensuring that the plasma–air–water system within remained closed, thus preventing any potential external fluxes which could perturb the experiment. The air gap, as measured from the bottom edge of the plastic casing to the water surface, was dg = 8.0 mm.
The powered electrode of the SBD reactor was square in shape with sides 40 mm in length. This electrode was placed on one side of a 2 mm thick quartz dielectric sheet, and on the opposite, liquid facing side of the quartz sheet the grounded electrode was attached. The ground electrode comprised of a hexagonal mesh, with each hexagon comprising of sides 3.6 mm in length and 0.7 mm in thickness. On application of a sufficiently high voltage, discharges formed along the sides of each hexagon, extending approximately 0.9 mm before terminating. Under the voltages considered in this study, no plasma was observed to form in the centre of each hexagon. This gave a plasma surface area of approximately Ap = 1 060 mm2 and an average plasma depth of dp = 0.2 mm.
The electrode was connected to a home-made power supply, capable of suppling voltages from 0 to 20 kV at 27 kHz. The voltage and current waveforms were monitored with an oscilloscope (Digital Phosphor, 500 MHz, 5GS/s, 4ch) and measured with a high voltage probe (TTP HVP 15 HF, 50 MHz, 30 kV, 1000:1) and a current monitor (Pearson model 2877).
Initial Water Parameters and Experimental Analysis
The water samples exposed to the plasma were provided by a water purification unit (Purite Analyst from Suez), which converts the tap water into a laboratory-quality purity (distilled water). The water obtained after purification had a low conductivity (2 ± 0.5 µS.m−1) and a pH at 6 ± 0.05.
The chemical analysis followed the techniques used to measure the water quality in a wastewater treatment plant. Those procedures have a high resolution and are preserved from any possible interference, which is the main problem in the quantification of species in the PAW. It was decided to focus on the evolution of 4 chemical compounds: nitrites (NO2−), nitrates (NO3−), nitrous acid (HNO2), and hydrogen ions (H+). An attempt was made to calculate the concentration of hydroxide ions (OH−) from the pH of the PAW, but the acidity of the solution meant that the concentrations would be so low as to be indistinguishable from numerical noise in the computations of the mathematical model, so it was abandoned.
The temperature of the PAW was measured directly during the experiment by a thermocouple immersed in the water (Signstek ST300 Dual Channel Digital Thermometer). The pH was measured right after the plasma exposure (HI-9813-6 pH/EC/TDS/°C with the probe HI-1285-6).
In all the experiments, the distilled water was used as a blank as it does not contain any compounds that might interfere with the chemical reagents.
Measurement of Aqueous Phase Species
The concentration of hydrogen ions ([H+]) was measured using the value of the pH obtained for the different exposure times and based on Eq. (1).
The concentration of nitrites [NO2−] and nitrous acid [HNO2] was determined using a colorimetric assay based on the Griess reagent [7, 25]. [HNO2] was estimated by the acid-basic relation between the nitrites and the nitrous acid, given by Eq. (2). This relation takes into consideration the value of the pH and the acidic constant of the couple (pKa1 = 3.39).
The concentration of nitrates [NO3−] was measured using a colorimetric assay based on the interaction of the nitrate ions with sodium salicylate (Sigma-Aldrich Ltd, CAS 54-21-7) in sulfuric acid medium after evaporation. The final complex has a yellow colouration observable in alkaline solution after the addition of sodium hydroxide. Revealed by spectrophotometry at 420 nm [26]. The concentration is obtained from a standard curve.
Model Description
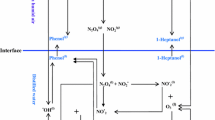
A global model was developed to describe the experimental set up outlined in Sect. 2. The model was defined over three distinct domains, as shown in Fig. 2a: a plasma region representing the luminous region observed in experiments, an air gap region filling the gap between the luminous plasma and the liquid’s surface; and a liquid region describing the water to be treated. All regions were modelled assuming spatially averaged densities with species fluxes being defined between any two adjacent regions. As the plasma was generated using an SBD, there was no direct contact between the plasma and the water, meaning that the plasma-generated species were transported to the water surface through the air gap region.

a Schematic diagram showing the types of species present and fluxes between each region, comparing the different gas–liquid flux terms for the one-film and two-film interface models. b Flowchart of the computational procedure
Computational Model Description
The model was essentially an upgraded version of that reported in [13, 20], as shown by the flow chart in Fig. 2b. Briefly, the model solved the conservation equation (Eq. (3)) for the densities of 53 species in the plasma region, which were governed by 625 reactions [27]. The model of the plasma and gas phases was based on an earlier experimentally-validated model of a similar discharge configuration, which was used to show that some of the species did not exist long enough to be transported into the air gap region while others did [13, 20]. As such, only the long-lived species (species that lived long enough to be transported into the gas region) were followed in the air gap region. The species which fall into each category are listed in the supplementary information.
The time-dependent continuity equation for the ith species in the plasma region was given by Eq. (3).
where Ci,p is the concentration of the ith species in the plasma phase (mol m−3); Ri,p is the overall plasma-phase reaction rate coefficient of the ith species (mol m−3 s−1); Γi,pg is the flux of the ith species from the plasma phase into the gas phase (mol m−2 s−1); and dp is the depth of the plasma phase (m).
The time-dependent continuity equation for the ith species in the air gap region was given by Eq. (4).
where Ci,g is the concentration of the ith species in the gas phase (mol m−3); Ri,g is the overall gas-phase reaction rate coefficient of the ith species (mol m−3 s−1); Γi,pg and Γi,gl are the fluxes of the ith species from the plasma phase and into the liquid phase respectively (mol m−2 s−1); and dg is the depth of the gas phase (m).
The final region was the liquid region, which consisted entirely of deionised water. Similar to the other two regions, the model solved the continuity equation for the concentrations of each of 34 aqueous species taking part in 139 reactions, given by Eq. (5). The full list of liquid-phase reactions with rate coefficients is found in the supplementary information [20, 22, 28,29,30,31,32,33].
where Ci,l is the concentration of the ith species in the liquid phase (mol m−3); Ri,l is the overall liquid-phase reaction rate coefficient of the ith species (mol m−3 s−1); Γi,gl is the flux of the ith species from the gas phase (mol m−2 s−1); and dl is the depth of the liquid phase (m).
The fluxes of the species were defined on the plasma-gas interface and on the gas–liquid interface. On the plasma-gas interface, the flux of each long-lived species was given by Eq. (6).
where Di,g is the gas-phase diffusion coefficient of the ith species (m2 s−1); Ci,p and Ci,g are the concentrations of the ith species in the plasma and gas regions respectively (mol m−3); and dp and dg are the depths of the plasma and gas regions respectively (m). To represent the gas–liquid interface, two theories were implemented: the one-film theory and the two-film theory; these are described in further detail in Sect. 3.2.
The parameters of the model were set to match those of the experiments. The air composition was assumed to be 78% N2, 21% O2 and 1% H2O with all plasma species assigned an initial number density of 5 × 106 m−3. At the beginning of the experiment, the water pH had been measured as 6 ± 0.05, which was input to the model as initial H+ and OH− concentrations of 1.0 × 10–3 mM and 1.0 × 10–5 mM respectively. As an increase in water temperature was observed during the experiment, a simple exponential curve was used to increase the temperature from 303 to 307 K through the course of the computation. The running time of the model was set to 1800s, which covered the duration of the experiment.
Modelling Transport Across the Gas–Liquid Interface
In both the one-film and the two-film theory, the equation used to describe the fluxes from the gas to the liquid phase and vice versa was given by Eq. (7) [34].
where HiCC is the dimensionless Henry’s solubility of the ith species, and Ri is the overall resistance to mass transfer of the ith species caused by the interface (s m−1).
The theories differ in how they calculate the resistance of the interface, as the one-film model must only account for one stagnant film, whereas the two-film model must account for two. The resistance factor in the one-film theory is given by Eq. (8).
where zf is the thickness of the stagnant film (m), and Di,w is the diffusion coefficient of the ith species in water (m2 s−1). In this work, the film is assumed to have a fixed thickness of zf = 1.0 mm. This value was chosen based on environmental studies that were supported by laboratory experiments to determine the flux of reactive and non-reactive gases across the air–water interface [35, 36].
To implement the two-film theory, the transport of species across the gas–liquid interface was also described by Eq. (7). The overall resistance of both stagnant films, Ri, is given by Eqs. (9)–(11) [11, 16,17,18].
where ki,l and ki,g represent the mass transfer coefficient of species i due to the stagnant film on the liquid and gas sides of the interface respectively, given by Eqs. (10)–(11) [16, 18].
where kO2,l is the liquid-phase mass transfer coefficient of O2, set in this work to a value of 4.0 × 10–6 m s−1 [16]; DO2,l is the liquid-phase diffusion coefficient of O2, set in this work to a value of 2 × 10–9 m2 s−1; Sh is the Sherwood number, discussed below; and L is a characteristic length, set in this work as the liquid depth, dl (= 4.5 mm).
As shown by Eq. (10), the liquid-phase mass transfer coefficient of any given species scales with its diffusion coefficient. This is because transport across the stagnant film is driven by molecular diffusion. If the mass-transfer and diffusion coefficients of any species are known, then the mass transfer coefficient of any other species with known diffusivity can be scaled accordingly [11, 37, 38]. While O2 was used as the reference species in this work, any other species can be substituted [16].
The Sherwood number is a dimensionless number used to describe the mass transfer coefficient through the stagnant film on the gas side of the gas–liquid interface. It is defined as the ratio of the rate of convective mass transfer in the stagnant film to that of diffusive mass transport in the gas–liquid interface. Since the existence of the film on the liquid side is already postulated in the one-film theory, the Sherwood number becomes very important in differentiating between the two models. Typical values of the Sherwood number in situations where the Reynold’s number is very small (and so there is only weak gas flow) are in the range of 6.58–17.90 [19]. In the absence of a known value of the Sherwood number for this scenario, a Sherwood number of Sh = 10 was used in this model as it has been determined to be around this order of magnitude that the Reynold’s number of fluid flow approaches zero.
Results and Discussion
Comparison Between Experiment and One Film Theory Model
The concentrations of the 4 aqueous species measured experimentally and compared with the predictions of the model assuming the one-film theory are shown in Fig. 3.

Predictions of the model (lines) with the interface modelled using 1-film theory compared with experimental data (points with error bars). Note that the line for [H+] almost exactly matches that for [NO3−] causing them to overlap
Experimentally, the two species with the highest concentration were H+ and NO3− with concentrations on the order of 10 mM. This agreed with the predictions of the one-film theory, but here the similarities end. The predicted concentrations of both H+ and NO3− initially rose at a much higher rate than either species did in the experiment, causing the computed concentrations to be higher than all of the experimental readings by at least an order of magnitude. Furthermore, [H+] and [NO3−] had clearly diverged in the experiment by the time of the reading at t = 20 min, whereas the one-film theory predicted that their concentrations would remain nearly identical throughout the whole experiment.
As H+ and NO3− are both ions, they are short-lived plasma species which are unable to diffuse from the plasma to the liquid surface, so they must be generated in the water by reactions of other species. Figure 4 shows the flux of the six species with the highest gas–liquid fluxes across the whole course of the computation, namely ozone (O3), nitric and nitrous acids (HNO3 and HNO2), hydrogen peroxide (H2O2), nitrous oxide (N2O) and dinitrogen pentoxide (N2O5). The flux of each of these species rose to a near-constant value within the first minute of the computation. Scrutiny of the reaction mechanism (see the supplementary information) shows that each of these species apart from H2O2 will readily react to produce both H+ and NO3− either directly or indirectly in the liquid phase.

Time evolution of the gas–liquid flux of the six long-lived plasma species with the highest flux, as predicted by computations using the one-film model
HNO3 dissociates upon dissolving in water, to produce only H+ and NO3−, while N2O5 reacts with water to produce the same, as shown in Eqs. (12) and (13).
According to the one-film model, these were the two species with the highest flux, so they were significant contributors to both the high concentrations of H+ and NO3− and the fact that their values were predicted to be always approximately equal.
The species with the next highest flux was HNO2 which also dissociates in aqueous solution, producing H+ and NO2− as shown in Eq. (14).
As reaction (14) produces H+ ions and no NO3− ions, this suggests that [H+] should rise higher than [NO3−]. There should also have been an increase in [NO2−] over time, whereas it remained relatively constant. However, there were two processes at work that prevented this from happening. Firstly, the flux of HNO3 was higher than that of HNO2 by factor of 10, and N2O5 was higher by a factor of 5, meaning the reactions of these two species dominated those of HNO2. Secondly, the aqueous NO2− ions produced are then involved in further reactions with high-flux species N2O and O3 as shown in Eqs. (15) and (16).
NO3− ions are a product of both reactions, helping to balance the [NO3−] with the increase in [H+] caused by the dissociation of HNO2. This also explains the approximately constant concentration of NO2− ions in the PAW predicted by the one-film theory, the magnitude of which showed good agreement with the experimental readings, although the measured concentration of NO2− showed a gradual decrease after time t = 15 min which was not seen in the results of the model.
The model predicted that the concentrations of H+, NO3− and HNO2 would rise in a near linear fashion throughout the experiment. While this agreed with the measurements of [HNO2], it was not true for [H+]—which showed an increasing rate of change—and [NO3−]—which showed a decreasing rate of change. This predicted linear behaviour can, however, be explained from the near-constant fluxes of the 6 dominant long-lived species combined with the reaction pathways explained above. As they are always being absorbed into the liquid bulk at a constant rate, the concentrations of any related aqueous species will likewise rise at a constant rate.
The patterns of results of this global model are comparable with those of models of similar experimental setups but modelling the liquid in one spatial dimension with the gas–liquid interface represented as a simple equilibrium law [20]. The one-film theory therefore allows purely global models to produce results of similar accuracy to dimensional models that require greater computing power and take vastly longer times to compute a solution.
Two-Film Interface Theory
To provide a comparison with the one-film theory, Fig. 5 shows the same experimental results as in Fig. 3 with the concentration of the 4 species against time as predicted by the model assuming the two-film theory.

Predictions of the model (lines) with the interface modelled using two-film theory compared with experimental data (points with error bars), plotted with the same scale as Figure 3 for ease of comparison. The value of the Sherwood number used in this case was Sh = 10
As in the experiment, the model assuming the two-film theory showed a divergence between the concentrations of H+ and NO3−, with [H+] immediately rising higher than [NO3−]. Although this differed from the experimental results, in which [NO3−] was lower than [H+] for at least the first reading at time t = 5 min, this was an improvement over the one-film theory which predicted that the concentrations of NO3− and H+ would be identical. A further improvement was seen in that, even though [NO3−] was still always overestimated as was [H+] for the first 5 readings until time t = 25 min, the predicted concentrations of both species were always much closer to the measured values than the results produced by the one-film theory.
Figure 6 shows the fluxes of the same 6 species across the experiment running time. By comparison with Fig. 4, it is apparent that the flux of HNO3 had dropped by at least a factor of 10 and N2O5 by at least a factor of 5, explaining the reduction in the concentrations of H+ and NO3− predicted by the two-film theory.

Time evolution of the gas–liquid flux of the six long-lived plasma species with the highest flux, as predicted by computations using the two-film theory, plotted with the same scale as Figure 4 for ease of comparison
The reduction in the flux of N2O5 was caused by a decrease in its gas-phase concentration, leading to a lower concentration gradient across the interface. There is only one reaction in the plasma- and gas-phase model that results in the production of N2O5, given here as Eq. (17) [13].
The gas-phase concentration of N2O5 was lower in the two-film theory because the concentration of gaseous NO3 was lower than in the one-film theory. This occurred because the equations for calculating flux across the gas–liquid interface in the two-film theory produced a smaller value for the mass transfer resistance than the corresponding value in the one-film theory as NO3 has a slightly hydrophilic nature.
The reason for the reduced flux of HNO3 is similar to the above reasoning for NO3, but greatly exaggerated as HNO3 is extremely hydrophilic. Indeed, it was by far the most hydrophilic of all the long-lived species included in this model, having a dimensionless Henry’s solubility several orders of magnitude higher than most other species [12]. As a result of the greatly decreased interfacial resistance in the two-film theory, equilibrium at the gas–liquid interface was achieved almost instantaneously. As a result, the concentration difference between the two phases remained small and so the flux remained much lower in the two-film theory than in the one-film theory.
HNO2 was the species with the highest flux in the two-film theory. Indeed, its flux doubled compared with the one-film theory, causing [HNO2] to be higher than the experimental readings by a factor of 10 at all times. At first this may seem a shortcoming of the two-film theory, however it should be noted that the one-film theory also overestimated [HNO2] by a factor of 6, so the overestimate in the two-film theory is not large and is justified by the great improvements in the accuracy of predictions of concentrations of H+ and NO3− ions.
The fact that HNO2 was the species with the highest flux in the two-film theory also explained the development of a difference between [H+] and [NO3−], which did not occur in the one-film theory as the flux of HNO2 was significantly less than the flux of HNO3 and N2O5, as explained above. This allowed the concentration of H+ ions to increase at a higher initial rate than NO3− ions due to reaction (14). The effect was made more obvious than in the one-film theory by the fact that the flux of HNO2 was about double that of the one-film theory. As well as an increased rate of H+ production compared to NO3− due to reaction (14), this effect also led to an increase in the rate of NO2− production, as is shown in Fig. 5 by relatively high concentration of NO2− in the two-film results. Compared with the one-film theory, [NO2−] was larger in the two-film theory by a factor of nearly 20.
The reason for the increased flux of HNO2 was the greatly decreased interfacial resistance in the two-film theory. This was the reason given above for the decreased flux of HNO3, but as shown previously in Eq. (14), HNO2 dissociates into H+ and NO2−. NO2− then goes on to react further (Eqs. (15) and (16)), producing NO3−, which contributes to the increased concentration of HNO3 whilst making sure that HNO2 never achieves an equilibrium, so a large concentration gradient is quickly established and continues throughout the whole computation.
The overestimates of [HNO2] and [NO3−] by an order of magnitude in the two-film theory are problems which both have the same root cause: the high flux of HNO2 across the gas–liquid interface. There are benefits to this high flux, as it has been shown in this work to have caused the difference between [H+] and [NO3] which was also observed in experiments. If the flux of HNO2 could be made to be lower in the two-film theory, while still being higher than the fluxes of HNO3 and N2O5 then that may solve the problem while keeping the benefits. One possible method of doing so is discussed in the next section.
Influence of the Sherwood Number
Considering that the Sherwood number is estimated experimentally, and it is not available for the exact experimental setup considered in this study, the model was run assuming the two-film theory with varying Sherwood number to evaluate its impact on the results. Four distinct cases were examined, with a Sherwood number of: Sh = 0.1, 1, 10 and 100, thus spanning a several orders of magnitude. The results, plotted on the same axes as the experimental results discussed previously, are shown in Fig. 7. It is clear that no specific value of the Sherwood number provides a perfect agreement with the experimental results for all species, with H+ and NO3− matching best to the profiles for higher Sherwood numbers and HNO2 and NO2− better matching the low Sherwood number cases.

The computed concentrations of a H+, b HNO2, c NO2− and d NO3− against time to show the effect of varying the Sherwood number in the two-film theory. Note that the curve for concentration of H+ and NO3− as predicted using the one-film theory have been scaled down by a factor of 10 as they quickly rise much higher than in the two-film theory for any value of the Sherwood number
It is reasonable to suspect that each species should be assigned its own Sherwood number, which could aid in addressing the differences between the predictions of the two-film theory and the experimental results. Since the Sherwood number is used in the two-film theory to estimate the thickness of the gas-phase stagnant film, this would imply that each species experiences a film of different thickness, and this has indeed been shown to be the case [39].
It is indicated by Fig. 7 that, if the correct value of the Sherwood number can be determined for each species, the two-film theory provided better agreement with experimental results than the one-film theory for most of the species measured. This is especially the case for H+ and NO3−, for which all tested values of the Sherwood number give closer agreement with the trend and magnitude of experimental concentrations than the one-film theory.
The two-film theory is therefore superior to the one-film theory in modelling mass transfer across the gas–liquid interface for the indirect generation of PAW by SBD. If an accurate value of the Sherwood number can be determined, even if only for the species with high flux, then the trends and magnitudes of species concentrations in the PAW can be determined with greater accuracy than the one-film theory. Even if the same value of the Sherwood number is used for all species, the most highly concentrated species show much better agreement with experiment than they do using the one-film theory. As the only difference between the two theories is the equation used to calculate flux across the interface, this improved accuracy comes with no extra cost in computing power or running time needed to compute a solution.
The main limiting factor in the accuracy of the two-film model is the lack of experimentally verified values for the Sherwood number assigned to each species. The Sherwood number of the ith species can be calculated by Eqs. (18)–(20) [16].
where Sci is the Schmidt number of the ith species; Re is the Reynolds number; ρ and μ are the mixture’s density (kg m−3) and viscosity (kg m−1 s−1) respectively; μi is the gaseous viscosity of the ith species (kg m−1 s−1); ρi is the partial density of the ith species in the gas phase (kg m−3); Dg,i is the diffusion coefficient of the ith species in the gas phase; L is the characteristic length as given in Eq. (11) (m); vi is the flow speed of the gaseous plasma species (m s−1).
Due to the high reactivity of most plasma generated species, there is very little experimentally validated data on the gaseous viscosity of these species and the errors associated with measuring their diffusivity in air are generally large. However, kinetic theory can be used to calculate theoretical values for these numbers, so a reasonable estimate of the Schmidt number of each species is feasible. The Reynolds number can be calculated by accurately coupling the momentum transport between the gas phase and the liquid phase.
Conclusions
Using a 0D computation model, this work has investigated the relative accuracies of applying the one-film and two-film theories of mass transfer across a gas–liquid interface to the generation of PAW by indirect treatment of an SBD air plasma source. It has demonstrated that such models are extremely sensitive to the theory used to describe the species fluxes across the gas–water interface. It has been shown that incorporating the two-film theory to describe the interfacial fluxes provides a significant improvement in the consistency between the predictions of the model of aqueous species concentration with those measured experimentally.
The two-film theory predicted the concentrations of the two dominant species, H+ and NO3− ions, to within a factor of 5 of that measured experimentally, whereas the one-film theory was not even accurate to within one order of magnitude. It also showed a difference between the concentrations of aqueous H+ and NO3− which was observed in experiment but not predicted by the model using the one-film theory. This was due to the two-film theory reducing the flux of HNO3 by a factor of 10 and N2O5 by a factor of 5, both of which dissociate to produce NO3− ions in water. This decrease in the flux N2O5 was due to a decrease in its density in the gas phase as the main reactant for its formation, NO3, was absorbed more easily in the two-film model. The lower flux of HNO3 was caused by an increase in the flux of the highly hydrophilic species HNO2 which began a series of reactions culminating in a rise in the rate of generation of HNO3 in the water, so the concentration difference across the interface was not as large as in the one-film model. However, the predicted increase in the flux of HNO2 led to an overestimate of the concentration of aqueous HNO2 by a factor of 10 and of aqueous NO2− ions by a factor of 20.
This work also investigated the influence of the Sherwood number on the computed concentrations of different species, it is shown that any value of the Sherwood number used within the range 0.1 ≤ Sh ≤ 100, which lies within realistic range for this parameter, yields more accurate results than the one-film theory in terms of both the value of the concentration of each species and its evolution over time. If each species is assigned its own value of the Sherwood number, this could solve the problem of overestimating the concentrations of aqueous HNO2 and NO2− without undoing the improvements in the concentrations of H+ and NO3−.
The main constraints on determining a value of the Sherwood number for each species are the difficulties in obtaining experimentally validated values of the Schmidt number of each species and the Reynolds number. While this is easily accomplished in the field of environmental science due to the stability of atmospheric gases, this is much more difficult in plasma science due to the high reactivity of many plasma generated species.
The results presented in this study provide an insight in to how best to model plasma-liquid interactions using a 0D global plasma model, providing a means to achieve an accurate representation of the aqueous phase chemistry without additional computational burden. As reactive species in the aqueous phase can be highly transient and often difficult to measure, such computational models will play an important part in supporting the development of future applications of PAW.
Data Availability
Data is available from the corresponding author upon reasonable request.
Code Availability
Commercial code COMSOL was used in this work.
References
Kamgang-Youbi G et al (2007) Evidence of temporal postdischarge decontamination of bacteria by gliding electric discharges: application to Hafnia alvei. Appl Environ Microbiol 73(15):4791–4796
Sivachandiran L, Khacef A (2017) Enhanced seed germination and plant growth by atmospheric pressure cold air plasma: combined effect of seed and water treatment. RSC Adv 7(4):1822–1832
Naumova IK, Maksimov AI, Khlyustova AV (2011) Stimulation of the germinability of seeds and germ growth under treatment with plasma-activated water. Surf Eng Appl Electrochem 47(3):263–265
Ma RN et al (2015) Non-thermal plasma-activated water inactivation of food-borne pathogen on fresh produce. J Hazard Mater 300:643–651
Sun P et al (2012) Inactivation of Bacillus subtilis spores in water by a direct-current, cold atmospheric-pressure air plasma microjet. Plasma Processes Polym 9(2):157–164
Locke BR et al (2006) Electrohydraulic discharge and nonthermal plasma for water treatment. Ind Eng Chem Res 45(3):882–905
Bruggeman PJ et al (2016) Plasma–liquid interactions: a review and roadmap. Plasma Sour Sci Technol 25(5):053002
Bruggeman P, Leys C (2009) Non-thermal plasmas in and in contact with liquids. J Phys D Appl Phys 42(5):053001
Heirman P, Van Boxem W, Bogaerts A (2019) Reactivity and stability of plasma-generated oxygen and nitrogen species in buffered water solution: a computational study. Phys Chem Chem Phys 21(24):12881–12894
Shiraiwa M et al (2012) Kinetic multi-layer model of gas-particle interactions in aerosols and clouds (KM-GAP): linking condensation, evaporation and chemical reactions of organics, oxidants and water. Atmos Chem Phys 12(5):2777–2794
Seader JD, Henley EJ, Roper DK (1998) Separation process principles, vol 25. Wiley, New York
Sander R (2015) Compilation of Henry’s law constants (version 4.0) for water as solvent. Atmos Chem Phys 15(8):4399–4981
Sakiyama Y et al (2012) Plasma chemistry model of surface microdischarge in humid air and dynamics of reactive neutral species. J Phys D Appl Phys 45(42):425201
Donelan, M.A.D., W. M.; Saltzman, E. S.; Wanninkhof, R. (2002) Gas transfer at water surfaces, American Geophysical Union
Wang C et al (2018) Beyond the standard two-film theory: computational fluid dynamics simulations for carbon dioxide capture in a wetted wall column. Chem Eng Sci 184:103–110
Guo Z, Roache N (2003) Overall mass transfer coefficient for pollutant emissions from small water pools under simulated indoor environmental conditions. Ann Occup Hyg 47(4):279–286
Meng F, Wen D, Sloan J (2008) Modelling of air-water exchange of PCBs in the Great Lakes. Atmos Environ 42(20):4822–4835
Qin N et al (2013) Atmospheric partitioning and the air-water exchange of polycyclic aromatic hydrocarbons in a large shallow Chinese lake (Lake Chaohu). Chemosphere 93(9):1685–1693
Colombet D et al (2013) Mass or heat transfer inside a spherical gas bubble at low to moderate Reynolds number. Int J Heat Mass Transf 67:1096–1105
Liu Z et al (2015) Physicochemical processes in the indirect interaction between surface air plasma and deionized water. J Phys D Appl Phys 48(49):495201
Liu ZC, Liu DX, Chen C, Liu ZJ, Yang AJ, Rong MZ, Chen HL, Kong MG (2018) Post-discharge evolution of reactive species in the water activated by a surface air plasma: a modeling study. J Phys D Appl Phys 51:175202
Lietz A, Kushner M (2016) Air plasma treatment of liquid covered tissue: long timescale chemistry. J Phys D Appl Phys 49(42):425205
Kruszelnicki J, Lietz A, Kushner M (2019) Atmospheric pressure plasma activation of water droplets. J Phys D Appl Phys 52(35):355207
Verlackt C, van Boxem W, Bogaerts A (2018) Transport and accumulation of plasma generated species in aqueous solution. Phys Chem Chem Phys 20:6845–6859
Bruggeman P et al (2016) Plasma-liquid interactions: a review and roadmap. Plasma Sour Sci Technol 25(5):053002
Rodier J (1975) Analysis of water. Wiley, New York City, NY, USA
Dickenson A et al (2018) The generation and transport of reactive nitrogen species from a low temperature atmospheric pressure air plasma source. Phys Chem Chem Phys 20(45):28499–28510
Herrmann H et al (2000) CAPRAM 2.3: A chemical aqueous phase radical mechanism for tropospheric chemistry. J Atmos Chem 36(3):231–284
Pandis S, Seinfeld J (1989) Sensitivity analysis of a chemical mechanism for aqueous-phase atmospheric chemistry. J Gerontol Ser A Biol Med Sci 94(D1):1105–1126
Pastina B, LaVerne J (2001) Effect of molecular hydrogen on hydrogen peroxide in water radiolysis. J Phys Chem A 105(40):9316–9322
Goldstein S, Lind J, Merenyi G (2005) Chemistry of peroxynitrites as compared to peroxynitrates. Chem Rev 105(6):2457–2470
Lukes P et al (2014) Aqueous-phase chemistry and bactericidal effects from an air discharge plasma in contact with water: evidence for the formation of peroxynitrite through a pseudo-second-order post-discharge reaction of H2O2 and HNO2. Plasma Sour Sci Technol 23(1):015019
Logager T, Sehested K (1993) Formation and decay of peroxynitrous acid—a pulse-radiolysis study. J Phys Chem 97(25):6664–6669
Basmadjian D (2004) Mass transfer: principles and applications. CRC Press, LLC, Boca Raton, FL, USA
Deacon EL (1977) Gas transfer to and across an air-water interface. Tellus 29(4):363–374
Bolin B (1960) On the exchange of carbon dioxide between the atmosphere and the Sea. Tellus 12(3):274–281
Jahne B, Haussecker H (1998) Air-water gas exchange. Annu Rev Fluid Mech 30:443–468
Portielje R, LiJklema L (1995) Carbon dioxide fluxes across the air-water interface and its impact on carbon availability in aquatic systems. Limnol Oceanogr 40(4):690–699
Pohl P, Saparov S, Antonenko Y (1998) The size of the unstirred layer as a function of the solute diffusion coefficient. Biophys J 75:1403–1409
Acknowledgements
The authors acknowledge the support of the Engineering and Physical Science Research Council (projects EP/S025790/1, EP/S026584/1, EP/R041849/1, and EP/T000104/1).
Funding
Engineering and Physical Science Research Council (projects EP/S025790/1, EP/S026584/1, EP/R041849/1, and EP/T000104/1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
There is no conflict of interest in this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Silsby, J.A., Simon, S., Walsh, J.L. et al. The Influence of Gas–Liquid Interfacial Transport Theory on Numerical Modelling of Plasma Activation of Water. Plasma Chem Plasma Process 41, 1363–1380 (2021). https://doi.org/10.1007/s11090-021-10182-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11090-021-10182-7