
Abstract
As one of the most common neuropathic disorders, neuropathic pain often has a negative impact on patients with persistent pain, mood disorders and sleep disturbances. Currently, neuropathic pain is not treated with any specific drug, instead, drugs for other diseases are used as replacements in clinics, but most have adverse effects. In recent years, the role of spinal cord microglia in the pathogenesis of neuropathic pain has been widely recognized, and they are being explored as potential therapeutic targets. Spinal microglia are known to be involved in the pathogenic mechanisms of neuropathic pain through purine signaling, fractalkine signaling, and p38 MAPK signaling. Exercise is a safe and effective treatment, and numerous studies have demonstrated its effectiveness in improving neurological symptoms. Nevertheless, it remains unclear what the exact molecular mechanism is. This review summarized the specific molecular mechanisms of exercise in alleviating neuropathic pain by mediating the activity of spinal microglia and maintaining the phenotypic homeostasis of spinal microglia through purine signaling, fractalkine signaling and p38 MAPK signaling. In addition, it has been proposed that different intensities and types of exercise affect the regulation of the above-mentioned signaling pathways differently, providing a theoretical basis for the improvement of neuropathic pain through exercise.
Similar content being viewed by others


Introduction
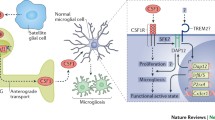
A major contributor to the global burden of disease, neuropathic pain (NPP) affects between 6.9% and 10% of the general population [1]. As well as persistent pain, neuropathic pain is often accompanied by mood disorders and sleep disturbances, which adversely affect patients’ quality of life [2, 3]. Currently, the primary treatment modalities for NPP are pharmacological. Opioids, cannabinoids, botulinum toxin, topical capsaicin, gabapentin, pregabalin, inhibitors of serotonin and noradrenalin reuptake, and tricyclic antidepressants (TCA) are recommended as first-line treatments according to evidence of high or moderate quality. TCAs inhibit neuronal hyperexcitability mainly by blocking Na+ and Ca2+ and adenosine activity [4]. However, TCAs can cause side effects such as dry mouth, confusion, urinary retention, and potential cardiotoxicity when taken for long periods of time [5]. The anticonvulsant carbamazepine decreases neuronal Na+ and Ca2+ inward flow, thereby reducing injurious stimulation of neurons [6], but the anticonvulsant carbamazepine also causes blurred vision, diplopia, nystagmus, and water intoxication when taken for long periods of time [7]. While several of these drugs have been developed for other indications (e.g. depression and epilepsy), there are no drugs that are specifically indicated for treating NPP. Additionally, these drugs have treatment-related adverse effects in the clinical phase. Meanwhile, these clinical drugs exert their therapeutic effects on NPP via Na+ and Ca2+ ion channels and do not regulate spinal microglia. Spinal cord microglia play a crucial role in the pathogenesis of neuropathic pain. However, the mechanisms underlying spinal microglial activation during neuropathic pain remain incompletely determined [8, 9]. Microglia are the first responders and principal resident immune cells of the central nervous system [10]. A large body of work has demonstrated that microglial activation is the early event that mediates neuroinflammation in neuropathic pain [11, 12]. Several studies have shown that exercise, as a systemic physical activity, can exert neuroprotective effects by strengthening neurogenesis and reducing the inflammatory response [13,14,15]. Although physical exercise has a neuroprotective effect by inhibiting microglia, fewer articles have investigated the relationship between exercise and spinal microglia in NPP. With the in-depth study of spinal microglia, a few signaling pathways, including fractalkine, purine signaling and others, are thought to be effective in regulating the expression of spinal microglia in NPP. In addition, the present study hypothesised the factors and signalling pathways involved in regulating NPP and generating neuroprotective effects through spinal microglia motility.
Microglia in Neuropathic Pain
A growing body of evidence indicates that spinal microglia react and undergo a series of changes that directly influence the establishment of neuropathic pain states [16]. After nerve damage, purinergic P2X4 receptors (nonselective cation channels activated by extracellular ATP) are upregulated in spinal microglia in a manner dependent on transcription factors interferon regulatory factor 8 and 5 (IRF8 and IRF5), both of which are expressed in microglia after peripheral nerve injury [17]. The expression of P2X4 receptors on the surface of microglia is regulated at the post-translational level by the CC chemokine receptor, chemotactic cytokine receptor 2. In addition, spinal microglia respond to extracellular stimuli through intracellular signaling pathways such as the mitogen-activated protein kinase, p38, and extracellular signal-regulated protein kinase pathways [18]. Inhibition of the function or expression of these microglial molecules suppresses the aberrant excitability of dorsal horn neurons and causes neuropathic pain [17].
Central nervous system (CNS) microglia are the main immunoreactive cells, which promote the proliferation and differentiation of neuronal cells and have a role in supporting, protecting and nourishing neurons [19,20,21,22]. Peripheral nerve injury activates microglia in the dorsal horn, which releases inflammatory mediators and pro-inflammatory mediators that sensitize neurons, leading to neuropathic pain [23, 24]. As a result, spinal microglia promote neuropathic pain by releasing several glial mediators that sensitize spinal neurons [25]. Meanwhile, microglia are regarded as a main source of inflammatory mediators (IFMs). These IFMs include interleukin-1 beta (IL-1β), interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), prostaglandin E2 (PGE2), nitric oxide, nerve growth factor and others. Spinal microglia activate nociceptive neurons in the CNS and peripheral nervous system (PNS), resulting in hypersensitivity to pain [26, 27]. NPP development was delayed by specific ablation of microglia, microglia produce pro-inflammatory cytokines such as IL-1β [28]. Studies have found that elevated IL-1β decreases the efficacy of morphine analgesia [29]. Also, inhibition of IL-1β can induce an anti-inflammatory response, thereby modulating microglial activation and subsequent pain hypersensitivity [28, 30, 31].
Therefore, according to these studies, spinal microglia play a significant role in the pathogenesis of NPP as well as its therapeutic mechanism. In the treatment of pain, it is expected to be a promising therapeutic target.
Exercise in Neuropathic Pain
Exercise, as a systemic physical activity, could be neuroprotective by strengthening neurogenesis and reducing inflammation. According to the current study, exercise reduces NPP in both animals and humans [32, 33]. It has been proven that spinal neuron excitability can be regulated by exercise training by increasing the synthesis of GAD-65 and GAD-67 in the dorsal horn of the spinal cord, as well as by reducing nerve potential after spinal cord injury and peripheral nerve injury [34]. An animal experiment found that in streptozotocin (STZ) rats, exercise alleviated mechanical hypersensitivity after four to five weeks. Meanwhile, this study also found that the expression of the mammalian target of rapamycin (mTOR) and IL-6 levels in rat dorsal root ganglions decreased. It seems to indicate that exercise regulates NPP through the mTOR pathway [35]. Moreover, the 5-week swim exercise reversed the increased expression of BDNF in DRG and decreased the up-regulation of NGF, normalized the phosphorylation of PLCg-1 on the dorsal side of spinal cord, and reversed astrocyte and microglia hyperactivity in the dorsal horn after nerve lesion [36]. In summary, exercise stimulates endogenous factors and signaling pathways that affect NPP.
The Mechanisms that Regulate Exercise to Reduce Neuropathic Pain
After exercise training, cytokines are released in the organism and promote increased synthesis of endogenous factors, such as brain-derived neurotrophic factor (BDNF), glutamic acid decarboxylase (GAD), tumor necrosis factor (TNF), interleukin et al. These factors alleviate NPP through relevant signaling pathways, including mTOR, transmembrane chemokine fractalkine, purine signaling, BDNF-tyrosine-protein kinase B (TrkB) and others [34, 35, 37, 38]. Evidence is now available to prove that these signaling pathways play an important role in NPP via spinal microglia. Additionally, exercise prevents microglia from becoming over-activated [39]. It has been shown that the effects of physical exercise on Alzheimer’s disease (AD) and Parkinson’s disease (PD) could reduce inflammatory processes, inhibit over-activation of microglial cells, and induce neuroprotective effects [13]. There is now evidence that exercise mediates microglia activation or maintains microglia phenotypic homeostasis by upregulating anti-inflammatory factor expression and downregulating pro-inflammatory factor expression. The effect of exercise on neurological symptoms may be mediated by microglia.
Exercise, Spinal Microglia and Neuropathic Pain
Exercise Plausible Downregulation of the Expression of Purine Signaling to Relieve Neuropathic Pain
The purinergic system is a critical regulatory pathway in the CNS and a basic signaling system that builds microglial behavior in different conditions. By expressing purinergic P2X and P2Y receptors in microglia, extracellular nucleotides (ADO, ATP, ADP, and AMP) play a critical role in neuron-microglia communication [40]. The microglial purinergic receptor plays a crucial role in neuropathic pain. Neuropathic pain was found to be exacerbated by P2X receptors (P2X4R and P2X7R) and P2Y receptors (P2Y6R, P2Y12R, P2Y13R, and P2Y14R). Activation of P2X/Y receptors can change intracellular molecular metabolism, activate related signaling pathways, release damaging factors, increase sensory information transmission, and trigger pain. It has been reported that sensitive P2X4R mediates increased BDNF and p38-MAPK for pain hypersensitivity [41]. According to studies in vivo, nicotinic antagonist treatment of hyperalgesia models up-regulates P2X4R in spinal microglia [41] and neuropathic pain models [42]. Additionally, blocking P2X4R pharmacologically reduces animals’ pain-related behaviors [43]. In a rat model of chronic constriction of the sciatic nerve (CCI), P2X7R expression is confined to microglia, and spinal microglia are activated after nerve injury [44]. BmK I-induced pain was associated with an increase in the expression of P2X7R in the ipsilateral spinal dorsal horn. The activation of microglial cells in the spinal dorsal horn is mediated by P2X7R up-regulation, which contributes to the development of pain [45]. The study shows that inhibition of p38 MAPK signaling suppressed the increase of P2Y6R, P2Y13R, and P2Y14R expression in microglia. A critical role for p38 MAPK signaling in NPP is played by regulating transcription of P2Y6R, P2Y13R, and P2Y14R in spinal microglia following nerve injury [46]. Both P2Y13R and P2Y12R are associated with microglia in the spinal cord that produce inflammatory cytokines, such as TNF-α, IL-1β, and IL-6 [47]. In vitro, P2Y13R and P2Y12R triggered paracrine signaling during inflammation [43, 48]. According to one study, P2Y12 and P2Y13 receptors regulate NPP synergistically. Nerve injuries increase the expression of P2Y13 receptors. The RhoA/ROCK pathway was activated in microglia, causing neuronal excitability and NPP [49]. A study has shown that the expression of P2Y12R mRNA was obviously increased in the spinal cord ipsilateral to the nerve injury and it was highly restricted to ionized-binding calcium adapter molecule 1-positive microglia expression. A P2Y13 receptor in the dorsal horn may also be involved in diabetic neuropathic pain. A vitro study found that co-culturing the N9 microglial line with a glioma cell line could upregulate P2Y14R expression, suggesting that P2Y14R may play a role in microglia-glioma communication [43, 50].
Currently, there are studies based on P2X and P2Y receptors as potential therapeutic targets. Research on the P2X and P2Y receptors as therapeutic targets for NPP is currently a hot area of research. It has been found that using electroacupuncture and receptor inhibitors can alleviate pain [51,52,53], but the intervention with exercise is rare. Exercise can regulate P2X and P2Y receptors expression and activation. In other diseases, P2X and P2Y receptors expression can be reduced by exercise to alleviate disease symptoms. The increases or decreases in receptor expression are related to the exercise type, intensity, frequency, and duration. A study has shown that moderate-intensity exercise can reduce the expression of P2X7R, thus regulating the relevant pathways to inflammation. However, low- and high-intensity exercise instead increased the expression of P2X7R [54]. Another study has shown that long-term moderate-intensity aerobic training may effectively reduce the occurrence of atherosclerotic thrombotic events by down-regulating the expression of P2Y12R [55]. Recently, P2X4R and P2X7R were speculated to be priming molecules in exercise-induced changes in BDNF, and a protective mechanism of exercise is provided in neurogenic diseases (including NPP). ATP is released from NPP-stimulated neurons and activates P2X4R and P2X7R in microglia via calcium channels. As purinoceptors become active, the p38 MAPK pathway is triggered, which releases TNF-α, IL-1β and BDNF [56]. Therefore, the regulation of P2X and P2Y receptor downregulation by exercise to alleviate NPP has become a meaningful research area. Choosing an appropriate exercise program is very important.
Exercise Plays a Double-Sided Role in Affecting Neuropathic Pain by Modulating Fractalkine on Spinal Microglia
C-X3-C motif chemokine ligand 1(CX3CL1, also known as fractalkine), a unique chemokine expressed by neurons constitutively, is a transmembrane chemokine that binds to the membrane. A receptor specific to fractalkine, CX3CR1(the receptor of CX3CL1), is mostly present on the surface of microglial cells within the dorsal spinal cord [57]. There is growing evidence that in the dorsal horn, fractalkine regulates neuron-microglia communication.
As a potential substrate for lysosomal cysteine protease Cathepsin S (CatS) cleavage, fractalkine plays a major role in CatS induction [58]. The CatS protein is essential for the activation of spinal microglia and NPP. The cysteine protease CatS from spinal microglial cells has previously been demonstrated to contribute to maintaining neuropathic hypersensitivity and activating microglia [58]. As we all know, NPP is promoted by the activation of spinal microglia. Thus, the activation of CatS has an induction role for NPP. Conversely, in the dorsal horn, the CatS inhibitor (morpholinurea-leucine-homophenylalaninevinylphenylsulfone, LHVS) attenuates microglia activation and NPP’s analgesic effect is reduced [58]. Research has demonstrated that neuronal soluble fractalkine (sFKN) is released by CatS pronociceptive effects and by CatS-induced microglia activation [25]. As a result of nerve injury, primary afferent fibers are activated, and mediators such as ATP are released, which causes the release of CatS from activated microglia, leading to the rapid release of sFKN. sFKN induces an increase in CX3CR1 that correlates with increased CX3CR1 expression on microglia due to nerve injury, and both together promote injury receptor hypersensitivity. A significant reduction in noxious stimulus responses could be caused by the loss of CX3CR1 [25]. In summary, in neuropathic conditions, increased primary afferent inputs to the dorsal horn cause microglial CatS release, which causes fractalkine liberation from dorsal horn neurons, which contributes to chronic pain amplification and maintenance through activation of CX3CR1 receptors on microglia. When fractalkine is activated on spinal microglia, p38 MAPK-mediated pathways are also activated [25, 58,59,60]. Due to the strong neuronal excitation in the spinal cord induced by upregulation of CX3CL1 during pathological pain conditions, the receptor expression of fractalkine is increased in microglia in pain-relevant areas [60]. Soluble secreted forms of fractalkine combined with CX3CR1 activate the proliferation and migration of microglia surrounding the affected area [25, 61]. Microglia contribute to enhanced nociceptive signaling when it comes to damaging stimuli to the peripheral nervous system [62]. Evidence shows that after PNI, microglia activation is induced by fractalkine signaling via CX3CR1. In response to p38 activation, proinflammatory cytokines such as TNF-a and IL-1 are synthesized [27]. Fractalkine signaling in the spine activates p38 phosphorylation, and there is a predominant expression of this process in spinal microglia in naive rats. After peripheral nerve injury, intra-cisternal administration of neutralizing antibodies against CX3CR1 could inhibit p38 phosphorylation. The NPP study shows the interaction between fractalkine and its microglial receptor (CX3CR1, P2X7R), which is critically involved in the maintenance of pain [60]. Interestingly, in contrast to its pro-injury effect in NPP, it has been shown that the neuronal fractalkine/microglial CX3CR1 system has a neuroprotective effect in diseases associated with neurodegeneration [63].
Consequently, inhibition of the fractalkine/CX3CR1 pathway may be an effective strategy for alleviating NPP. However, there is variability in the modulatory effects of different exercises on fractaline. Fractalkine expression was significantly upregulated in resistance and acute exercise. A previous study found that the fractalkine mRNA of eight untrained men was significantly elevated 2 h post-intense resistance exercise [64]. Meanwhile, a clinical study found that 12 cytokines and myokines showed significant alterations after low-intensity resistance exercise compared to before exercise. It was found that Fractalkine/CX3CL1 was the most significantly upregulated by exercise [65]. In addition, recent studies have demonstrated that CX3CR1 expression is induced in the spinal cord by this substance during intense acute swimming [66]. However, in a study of aerobic exercise, researchers found that aerobic exercise reduces plasma levels of fractalkine in younger adults [67]. The results of these studies suggest that exercise may regulate fractalin in response to inflammation. Acute exercise and resistance exercise trigger inflammation, while regular aerobic exercise suppresses inflammation [68]. As a result, different exercise intensities and types may have different effects on the expression of NPP.
As a result, we suggested that exercise regulates the fractalkine pathway through spinal microglia. The expression of fractalkine is associated with the pathogenic mechanism of NPP. In this respect, our campaign may suggest that improving NPP via fractalkine is an issue that should be explored further.
Exercise Downregulates p38 MAPK Signaling, Reducing the Activation of Spinal Microglia and the Generation of Hyperalgesia via Spinal Microglia
Mitogen-activated protein kinases (MAPKs) are a family of evolutionarily conserved molecules that play an important role in cell signaling and gene expression. There are three major members of the MAPK family, representing three different signaling cascades that are known to participate in the generation of hyperalgesia. These members include extracellular signal-regulated kinase (ERK), p38, and c-Jun N-terminal kinase (JNK) [69, 70]. The p38 MAPK is regarded as a stress-induced kinase, and it plays a critical role in inflammation. The inhibitor of p38 has been shown to effectively relieve rheumatoid arthritis and inflammatory pain [71].
Numerous studies have demonstrated that p38 activation in spinal microglia plays a significant role in NPP. The antibody of phosphorylated p38 was used in a study to examine activation of p38 in the rat spinal cord after spinal nerve ligation (SNL), a model commonly used to study NPP. This result indicated that the increase in phosphorylation of p38 is accompanied by an increase in p38 activity, which means ATF-2, a substrate of p38, is phosphorylated at a higher level. Meanwhile, p38 is activated in spinal cells labeled with the microglial marker CD11b [26, 27, 71, 72]. Additionally, recent studies have shown that the application of p38 inhibitors such as FR167653, CNI-1493 and SB203580 can reverse early NPP [27, 71].
There is no doubt that p38 is an important signaling factor for microglia. In recent years, several studies have investigated the upstream mechanisms that cause the activation of p38 by spinal microglia in NPP [9, 71, 73]. It was proven that NPP is associated with p38 MAPK activation and the accumulation of inflammatory cytokines in the spinal cord. As mentioned above, several inflammatory factors, such as IL-1β, IL-6 and TNF-α, are released by nerve spontaneous activity after nerve injury. It is currently confirmed that TNF-α inhibitors prevent NPP development by inhibiting spinal p38 activation [72]. Meanwhile, the hyperalgesia, which is produced by IL-1β and substance P, could be abolished by a p38 inhibitor [74, 75]. Additionally, according to recent research, chemokines have been shown to be important mediators of neural-microglial interactions and the sensitization of NPP [60]. Fractalkine/CX3CR1, a typical chemokine, is activated by CatS on spinal cord microglia following nerve injury, thereby enhancing NPP [58]. Therefore, the development of NPP requires the fractalkine-CX3CR1-p38 cascade. The purinergic system has also been shown to activate p38 by activating receptors on the spinal microglia in NPP. With the continuous peripheral noxious stimulation, maybe the microglial P2X7R is responsible for the release of the sensory neuronal fractalkine, which stimulates the phosphorylation of p38 MAPK in microglia, thereby activating neurons through the release of pronociceptive mediators [76, 77].
Thus, p38 MAPK in spinal microglia is activated by the above 3 pathways. In the downstream mechanism of p38 MAPK, p38 activates phospholipase A2 (PLA2) through its downstream kinase MAPK-activated protein kinase-2 (MAPKAP-2), and after activating PLA2, arachidonic acid is produced for the generation of prostaglandins, which are then catalyzed by COX to produce PGE2 [78]. As previously mentioned, posttranslational regulation by P38 can also control the synthesis of IFMs. Activation of p38 increases the expression of inflammatory mediators and growth factors (including cytokines and BDNF) which are secreted via the transcription factor NF-κB. With the release of these mediators, nociceptive dorsal horn neurons are sensitized via both presynaptic and postsynaptic mechanisms, which leads to persistent hyperalgesia [26].
While there is no clear evidence that exercise can improve NPP through p38 MAPK, research has confirmed that exercise can have neuroprotective effects through p38 MAPK [39, 79]. Hyperalgesia and the decrease of the pain threshold for nociceptive stimulation are the most common manifestations of NPP. P38 phosphorylation plays a key role in the formation of hyperalgesia and tolerance by releasing a multitude of inflammatory mediators from the microglia, including IL-1β, IL-6, and TNF-α [27].
An experiment conducted on mice with morphine-induced hyperalgesia found that running at a speed of 20 m per minute for 30 min every day reduced the development of morphine hyperalgesia and tolerance, and exercise reduced p-p38 expression in the presence of morphine. This is consistent with the results of applying p38 inhibitors (SB203580) [39]. Additionally, this study also used the same exercise protocol to exercise precondition rats after simulated neonatal incision surgery-induced enhanced hyperalgesia and also found that exercise preconditioning reduced hyperalgesia [79]. It has been reported that exercise mediates inflammation in the central nervous system, prevents microglia activation, and has antinociceptive effects [39]. As a result of reduced inflammation and inhibition of microglial over activity, p38 phosphorylation is reduced, resulting in an increase in nociceptive thresholds and an improvement in hypersensitivity. There is now evidence that exercise can reduce nociceptive hypersensitivity via the p38 MAPK pathway, while p38 MAPK plays a key role in NPP. Therefore, whether the mechanism by which exercise improves NPP is related to the p38 MAPK pathway is an issue worth exploring in the future.
Conclusion
It is proven that purine signaling, fractalkine signaling, and p38 MAPK signaling play a significant role in pathological processes affecting NPP via spinal microglia (Table 1). Exercise modulates the p38 MAPK pathway to improve hyperalgesia and produce neuroprotective effects. However, there is limited efficacy in clinical trials based on the current research. Fractalkine and purine are able to affect the NPP, and exercise modulates their expression through spinal microglia. With this evidence it can be hypothesised that exercise can affect the development of NPP by regulating spinal microglia. In addition, the latest in vivo experiment found that exercise enhanced autophagy through the BDNF/ AKT/mTOR pathway and promoted m1-m2 polarization of microglia to improve neuropathic pain. Again, this implies that exercise may improve the path. Furthermore, different intensities and types of exercise, such as moderate-intensity exercise, acute exercise, resistance exercise, and aerobic exercise contribute to the down-regulation of purine receptors, the p38 MAPK pathway, and the simultaneous upward and downregulation of fractalkine. Considering these findings, we speculate that more clinical studies would be necessary to clarify the relationship between the type and intensity of exercise and NPP, as well as its underlying mechanisms in the future.
Data Availability
Not applicable.
Code Availability
Not applicable.
Abbreviations
- AD:
-
Alzheimer’s disease
- BDNF:
-
Brain-derived neurotrophic factor
- CatS:
-
Cathepsin S
- CCI:
-
A rat model of chronic constriction of the sciatic nerve
- CNS:
-
Central nervous system
- CX3CL1/fractalkine:
-
C-X3-C motif chemokine ligand 1
- ERK:
-
Extracellular signal regulated kinase
- GAD:
-
Glutamic acid decarboxylase
- IFMs:
-
Inflammatory mediators
- IL-1β:
-
Interleukin-1 beta
- IL-6:
-
Interleukin-6
- JNK:
-
C-Jun N-terminal kinase
- LHVS:
-
Morpholinurea-leucine-homophenylalanine-vinylsulfone phenyl
- MAPK:
-
Mitogen-activated protein kinases
- MAPKAP-2:
-
MAPK activated protein kinase-2
- mTOR:
-
Mammalian target of rapamycin
- NPP:
-
Neuropathic pain
- PD:
-
Parkinson’s disease
- PGE2:
-
Prostaglandin E2
- PNS:
-
Peripheral nervous system
- sFKN:
-
Soluble fractalkine
- SNL:
-
Spinal nerve ligation
- STZ:
-
Streptozotocin
- TCA:
-
Tricyclic antidepressants
- TNF:
-
Tumor necrosis factor
- TNF-α:
-
Tumor necrosis factor-alpha
- TrkB:
-
BDNF-tyrosine-protein kinase B
- PLA2:
-
Phospholipase A2
References
Scholz J, Finnerup NB, Attal N, Aziz Q, Baron R, Bennett MI, Benoliel R, Cohen M, Cruccu G, Davis KD, Evers S, First M, Giamberardino MA, Hansson P, Kaasa S, Korwisi B, Kosek E, Lavand’homme P, Nicholas M, Nurmikko T, Perrot S, Raja SN, Rice ASC, Rowbotham MC, Schug S, Simpson DM, Smith BH, Svensson P, Vlaeyen JWS, Wang SJ, Barke A, Rief W, Treede RD (2019) The IASP classification of chronic pain for ICD-11: chronic neuropathic pain. Pain 160(1):53–59. https://doi.org/10.1097/j.pain.0000000000001365
Giovannini S, Coraci D, Brau F, Galluzzo V, Loreti C, Caliandro P, Padua L, Maccauro G, Biscotti L, Bernabei R (2021) Neuropathic pain in the elderly. Diagnostics (Basel). https://doi.org/10.3390/diagnostics11040613
Serrano Afonso A, Carnaval T, Videla Cés S (2021) Combination therapy for neuropathic pain: a review of recent evidence. J Clin Med. https://doi.org/10.3390/jcm10163533
Miller A, Rabe-Jabłońska J (2005) The effectiveness of antidepressants in the treatment of chronic non-cancer pain–a review. Psychiatr Pol 39(1):21–32
Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, Gilron I, Haanpää M, Hansson P, Jensen TS, Kamerman PR, Lund K, Moore A, Raja SN, Rice AS, Rowbotham M, Sena E, Siddall P, Smith BH, Wallace M (2015) Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol 14(2):162–173. https://doi.org/10.1016/s1474-4422(14)70251-0
Freynhagen R, Strojek K, Griesing T, Whalen E, Balkenohl M (2005) Efficacy of pregabalin in neuropathic pain evaluated in a 12-week, randomised, double-blind, multicentre, placebo-controlled trial of flexible- and fixed-dose regimens. Pain 115(3):254–263. https://doi.org/10.1016/j.pain.2005.02.032
Hurley RW, Chatterjea D, Rose Feng M, Taylor CP, Hammond DL (2002) Gabapentin and pregabalin can interact synergistically with naproxen to produce antihyperalgesia. Anesthesiology 97(5):1263–1273. https://doi.org/10.1097/00000542-200211000-00033
Tsuda M (2016) Microglia in the spinal cord and neuropathic pain. J Diabetes Investig 7(1):17–26. https://doi.org/10.1111/jdi.12379
Wang L, Yin C, Liu T, Abdul M, Zhou Y, Cao JL, Lu C (2020) Pellino1 regulates neuropathic pain as well as microglial activation through the regulation of MAPK/NF-κB signaling in the spinal cord. J Neuroinflammation 17(1):83. https://doi.org/10.1186/s12974-020-01754-z
Salter MW, Beggs S (2014) Sublime microglia: expanding roles for the guardians of the CNS. Cell 158(1):15–24. https://doi.org/10.1016/j.cell.2014.06.008
Mika J, Osikowicz M, Rojewska E, Korostynski M, Wawrzczak-Bargiela A, Przewlocki R, Przewlocka B (2009) Differential activation of spinal microglial and astroglial cells in a mouse model of peripheral neuropathic pain. Eur J Pharmacol 623(1–3):65–72. https://doi.org/10.1016/j.ejphar.2009.09.030
Tanga FY, Raghavendra V, DeLeo JA (2004) Quantitative real-time RT-PCR assessment of spinal microglial and astrocytic activation markers in a rat model of neuropathic pain. Neurochem Int 45(2–3):397–407. https://doi.org/10.1016/j.neuint.2003.06.002
de Almeida EJR, Ibrahim HJ, Chitolina Schetinger MR, de Andrade CM, Cardoso AM (2022) Modulation of inflammatory mediators and microglial activation through physical exercise in Alzheimer’s and Parkinson’s diseases. Neurochem Res 47(11):3221–3240. https://doi.org/10.1007/s11064-022-03713-x
Liu MX, Luo L, Fu JH, He JY, Chen MY, He ZJ, Jia J (2022) Exercise-induced neuroprotection against cerebral ischemia/reperfusion injury is mediated via alleviating inflammasome-induced pyroptosis. Exp Neurol 349:113952. https://doi.org/10.1016/j.expneurol.2021.113952
Angulo J, El Assar M, Álvarez-Bustos A, Rodríguez-Mañas L (2020) Physical activity and exercise: strategies to manage frailty. Redox Biol 35:101513. https://doi.org/10.1016/j.redox.2020.101513
Tsuda M, Masuda T, Tozaki-Saitoh H, Inoue K (2013) P2X4 receptors and neuropathic pain. Front Cell Neurosci 7:191. https://doi.org/10.3389/fncel.2013.00191
Wei TH, Hsieh CL (2020) Effect of acupuncture on the p38 signaling pathway in several nervous system diseases: a systematic review. Int J Mol Sci. https://doi.org/10.3390/ijms21134693
Qin J, Ma Z, Chen X, Shu S (2023) Microglia activation in central nervous system disorders: A review of recent mechanistic investigations and development efforts. Front Neurol 14:1103416. https://doi.org/10.3389/fneur.2023.1103416
Mapplebeck JCS, Beggs S, Salter MW (2016) Sex differences in pain: a tale of two immune cells. Pain 157(Suppl 1):S2-s6. https://doi.org/10.1097/j.pain.0000000000000389
Hendriksen E, van Bergeijk D, Oosting RS, Redegeld FA (2017) Mast cells in neuroinflammation and brain disorders. Neurosci Biobehav Rev 79:119–133. https://doi.org/10.1016/j.neubiorev.2017.05.001
Stoyanov S, Sun W, Düsedau HP, Cangalaya C, Choi I, Mirzapourdelavar H, Baidoe-Ansah D, Kaushik R, Neumann J, Dunay IR, Dityatev A (2021) Attenuation of the extracellular matrix restores microglial activity during the early stage of amyloidosis. Glia 69(1):182–200. https://doi.org/10.1002/glia.23894
Kohno K, Shirasaka R, Yoshihara K, Mikuriya S, Tanaka K, Takanami K, Inoue K, Sakamoto H, Ohkawa Y, Masuda T, Tsuda M (2022) A spinal microglia population involved in remitting and relapsing neuropathic pain. Science 376(6588):86–90. https://doi.org/10.1126/science.abf6805
Weng HR, Gao M, Maixner DW (2014) Glycogen synthase kinase 3 beta regulates glial glutamate transporter protein expression in the spinal dorsal horn in rats with neuropathic pain. Exp Neurol 252:18–27. https://doi.org/10.1016/j.expneurol.2013.11.018
Serizawa K, Tomizawa-Shinohara H, Miyake S, Yogo K, Matsumoto Y (2021) Interleukin-6: evolving role in the management of neuropathic pain in neuroimmunological disorders. Inflamm Regen 41(1):34. https://doi.org/10.1186/s41232-021-00184-5
Clark AK, Yip PK, Malcangio M (2009) The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. J Neurosci 29(21):6945–6954. https://doi.org/10.1523/jneurosci.0828-09.2009
Tsuda M, Mizokoshi A, Shigemoto-Mogami Y, Koizumi S, Inoue K (2004) Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia 45(1):89–95. https://doi.org/10.1002/glia.10308
Ji RR, Suter MR (2007) p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain 3:33. https://doi.org/10.1186/1744-8069-3-33
Yi MH, Liu YU, Liu K, Chen T, Bosco DB, Zheng J, Xie M, Zhou L, Qu W, Wu LJ (2021) Chemogenetic manipulation of microglia inhibits neuroinflammation and neuropathic pain in mice. Brain Behav Immun 92:78–89. https://doi.org/10.1016/j.bbi.2020.11.030
Pilat D, Rojewska E, Jurga AM, Piotrowska A, Makuch W, Przewlocka B, Mika J (2015) IL-1 receptor antagonist improves morphine and buprenorphine efficacy in a rat neuropathic pain model. Eur J Pharmacol 764:240–248. https://doi.org/10.1016/j.ejphar.2015.05.058
Sharif K, Watad A, Bragazzi NL, Lichtbroun M, Amital H, Shoenfeld Y (2018) Physical activity and autoimmune diseases: get moving and manage the disease. Autoimmun Rev 17(1):53–72. https://doi.org/10.1016/j.autrev.2017.11.010
Pan C, Wang C, Zhang L, Song L, Chen Y, Liu B, Liu WT, Hu L, Pan Y (2018) Procyanidins attenuate neuropathic pain by suppressing matrix metalloproteinase-9/2. J Neuroinflammation 15(1):187. https://doi.org/10.1186/s12974-018-1182-9
Bobinski F, Teixeira JM, Sluka KA, Santos ARS (2018) Interleukin-4 mediates the analgesia produced by low-intensity exercise in mice with neuropathic pain. Pain 159(3):437–450. https://doi.org/10.1097/j.pain.0000000000001109
Leitzelar BN, Koltyn KF (2021) Exercise and neuropathic pain: a general overview of preclinical and clinical research. Sports Med Open 7(1):21. https://doi.org/10.1186/s40798-021-00307-9
Li X, Wang Q, Ding J, Wang S, Dong C, Wu Q (2020) Exercise training modulates glutamic acid decarboxylase-65/67 expression through TrkB signaling to ameliorate neuropathic pain in rats with spinal cord injury. Mol Pain 16:1744806920924511. https://doi.org/10.1177/1744806920924511
Ma X, Liu S, Liu D, Wang Q, Li H, Zhao Z (2020) Exercise intervention attenuates neuropathic pain in diabetes via mechanisms of mammalian target of rapamycin (mTOR). Arch Physiol Biochem 126(1):41–48. https://doi.org/10.1080/13813455.2018.1489851
Almeida C, DeMaman A, Kusuda R, Cadetti F, Ravanelli MI, Queiroz AL, Sousa TA, Zanon S, Silveira LR, Lucas G (2015) Exercise therapy normalizes BDNF upregulation and glial hyperactivity in a mouse model of neuropathic pain. Pain 156(3):504–513. https://doi.org/10.1097/01.j.pain.0000460339.23976.12
Chuganji S, Nakano J, Sekino Y, Hamaue Y, Sakamoto J, Okita M (2015) Hyperalgesia in an immobilized rat hindlimb: effect of treadmill exercise using non-immobilized limbs. Neurosci Lett 584:66–70. https://doi.org/10.1016/j.neulet.2014.09.054
Sluka KA, Frey-Law L, Hoeger Bement M (2018) Exercise-induced pain and analgesia? Underlying mechanisms and clinical translation. Pain 159(1):S91–S97. https://doi.org/10.1097/j.pain.0000000000001235
Gong X, Fan R, Zhu Q, Ye X, Chen Y, Zhang M (2021) Exercise reduces morphine-induced hyperalgesia and antinociceptive tolerance. Biomed Res Int 2021:6667474. https://doi.org/10.1155/2021/6667474
Tsuda M, Inoue K (2016) Neuron-microglia interaction by purinergic signaling in neuropathic pain following neurodegeneration. Neuropharmacology 104:76–81. https://doi.org/10.1016/j.neuropharm.2015.08.042
Zhang X, Xu P, Li C, Zhu W, Wu S, Yu A, Ding Y, Wang Q, Zhang Z (2017) Spinal microglial P2X4 receptor-brain-derived neurotrophic factor signaling regulates nicotine withdrawal-induced hyperalgesia. NeuroReport 28(6):339–347. https://doi.org/10.1097/wnr.0000000000000769
Jurga AM, Piotrowska A, Makuch W, Przewlocka B, Mika J (2017) Blockade of P2X4 receptors inhibits neuropathic pain-related behavior by preventing MMP-9 activation and consequently, pronociceptive interleukin release in a rat model. Front Pharmacol 8:48. https://doi.org/10.3389/fphar.2017.00048
Calovi S, Mut-Arbona P, Sperlágh B (2019) Microglia and the purinergic signaling system. Neuroscience 405:137–147. https://doi.org/10.1016/j.neuroscience.2018.12.021
He WJ, Cui J, Du L, Zhao YD, Burnstock G, Zhou HD, Ruan HZ (2012) Spinal P2X(7) receptor mediates microglia activation-induced neuropathic pain in the sciatic nerve injury rat model. Behav Brain Res 226(1):163–170. https://doi.org/10.1016/j.bbr.2011.09.015
Zhou J, Zhang X, Zhou Y, Wu B, Tan ZY (2019) Up-regulation of P2X7 receptors contributes to spinal microglial activation and the development of pain induced by BmK-I. Neurosci Bull 35(4):624–636. https://doi.org/10.1007/s12264-019-00345-0
Kobayashi K, Yamanaka H, Yanamoto F, Okubo M, Noguchi K (2012) Multiple P2Y subtypes in spinal microglia are involved in neuropathic pain after peripheral nerve injury. Glia 60(10):1529–1539. https://doi.org/10.1002/glia.22373
Liu PW, Yue MX, Zhou R, Niu J, Huang DJ, Xu T, Luo P, Liu XH, Zeng JW (2017) P2Y(12) and P2Y(13) receptors involved in ADPβs induced the release of IL-1β, IL-6 and TNF-α from cultured dorsal horn microglia. J Pain Res 10:1755–1767. https://doi.org/10.2147/jpr.S137131
Quintas C, Vale N, Gonçalves J, Queiroz G (2018) Microglia P2Y(13) receptors prevent astrocyte proliferation mediated by P2Y(1) receptors. Front Pharmacol 9:418. https://doi.org/10.3389/fphar.2018.00418
Tatsumi E, Yamanaka H, Kobayashi K, Yagi H, Sakagami M, Noguchi K (2015) RhoA/ROCK pathway mediates p38 MAPK activation and morphological changes downstream of P2Y12/13 receptors in spinal microglia in neuropathic pain. Glia 63(2):216–228. https://doi.org/10.1002/glia.22745
Curet MA, Watters JJ (2018) P2Y14 receptor activation decreases interleukin-6 production and glioma GL261 cell proliferation in microglial transwell cultures. J Neurooncol 137(1):23–31. https://doi.org/10.1007/s11060-017-2700-9
Yang H, Wang L, Chu X, Shi X, Li X, Li T (2022) BoNT/A alleviates neuropathic pain in osteoarthritis by down-regulating the expression of P2X4R in spinal microglia. Toxicon 206:55–63. https://doi.org/10.1016/j.toxicon.2021.12.012
Wu Q, Yue J, Lin L, Yu X, Zhou Y, Ying X, Chen X, Tu W, Lou X, Yang G, Zhou K, Jiang S (2021) Electroacupuncture may alleviate neuropathic pain via suppressing P2X7R expression. Mol Pain 17:1744806921997654. https://doi.org/10.1177/1744806921997654
Zhou R, Xu T, Liu X, Chen Y, Kong D, Tian H, Yue M, Huang D, Zeng J (2018) Activation of spinal dorsal horn P2Y(13) receptors can promote the expression of IL-1β and IL-6 in rats with diabetic neuropathic pain. J Pain Res 11:615–628. https://doi.org/10.2147/jpr.S154437
Li Z, Huang Z, Zhang H, Lu J, Tian Y, Piao S, Lin Z, Bai L (2021) Moderate-intensity exercise alleviates pyroptosis by promoting autophagy in osteoarthritis via the P2X7/AMPK/mTOR axis. Cell Death Discov 7(1):346. https://doi.org/10.1038/s41420-021-00746-z
Taghizadeh M, Kargarfard M, Braune S, Jung F, Naderi M (2022) Long-term aerobic exercise training in type two diabetic patients alters the expression of miRNA-223 and its corresponding target, the P2RY12 receptor, attenuating platelet function. Clin Hemorheol Microcirc 80(2):107–116. https://doi.org/10.3233/ch-211209
Sun BX, Peng AS, Liu PJ, Wang MJ, Ding HL, Hu YS, Kang L (2023) Neuroprotection of exercise: P2X4R and P2X7R regulate BDNF actions. Purinergic Signal 19(1):297–303. https://doi.org/10.1007/s11302-022-09879-x
Ni CM, Ling BY, Xu X, Sun HP, Jin H, Zhang YQ, Cao H, Xu L (2020) CX3CR1 contributes to streptozotocin-induced mechanical allodynia in the mouse spinal cord. J Zhejiang Univ Sci B 21(2):166–171. https://doi.org/10.1631/jzus.B1900439
Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, Dehvari M, Wotherspoon G, Winter J, Ullah J, Bevan S, Malcangio M (2007) Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci U S A 104(25):10655–10660. https://doi.org/10.1073/pnas.0610811104
Staniland AA, Clark AK, Wodarski R, Sasso O, Maione F, D’Acquisto F, Malcangio M (2010) Reduced inflammatory and neuropathic pain and decreased spinal microglial response in fractalkine receptor (CX3CR1) knockout mice. J Neurochem 114(4):1143–1157. https://doi.org/10.1111/j.1471-4159.2010.06837.x
Clark AK, Malcangio M (2014) Fractalkine/CX3CR1 signaling during neuropathic pain. Front Cell Neurosci 8:121. https://doi.org/10.3389/fncel.2014.00121
Ji RR, Chamessian A, Zhang YQ (2016) Pain regulation by non-neuronal cells and inflammation. Science 354(6312):572–577. https://doi.org/10.1126/science.aaf8924
Eyo UB, Peng J, Murugan M, Mo M, Lalani A, Xie P, Xu P, Margolis DJ, Wu LJ (2016) Regulation of physical microglia-neuron interactions by fractalkine signaling after status epilepticus. eNeuro. https://doi.org/10.1523/eneuro.0209-16.2016
Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM (2006) Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 9(7):917–924. https://doi.org/10.1038/nn1715
Della Gatta PA, Cameron-Smith D, Peake JM (2014) Acute resistance exercise increases the expression of chemotactic factors within skeletal muscle. Eur J Appl Physiol 114(10):2157–2167. https://doi.org/10.1007/s00421-014-2936-4
Hashida R, Matsuse H, Kawaguchi T, Yoshio S, Bekki M, Iwanaga S, Sugimoto T, Hara K, Koya S, Hirota K, Nakano D, Tsutsumi T, Kanto T, Torimura T, Shiba N (2021) Effects of a low-intensity resistance exercise program on serum miR-630, miR-5703, and Fractalkine/CX3CL1 expressions in subjects with No exercise habits: A preliminary study. Hepatol Res 51(7):823–833. https://doi.org/10.1111/hepr.13670
Borghi SM, Bussulo SKD, Pinho-Ribeiro FA, Fattori V, Carvalho TT, Rasquel-Oliveira FS, Zaninelli TH, Ferraz CR, Casella AMB, Cunha FQ, Cunha TM, Casagrande R, Verri WA Jr (2021) Intense acute swimming induces delayed-onset muscle soreness dependent on spinal cord neuroinflammation. Front Pharmacol 12:734091. https://doi.org/10.3389/fphar.2021.734091
Kumar P, Stiernborg M, Fogdell-Hahn A, Månsson K, Furmark T, Berglind D, Melas PA, Forsell Y, Lavebratt C (2022) Physical exercise is associated with a reduction in plasma levels of fractalkine, TGF-β1, eotaxin-1 and IL-6 in younger adults with mobility disability. PLoS One 17(2):e0263173. https://doi.org/10.1371/journal.pone.0263173
Qu H, Liu R, Chen J, Zheng L, Chen R (2020) Aerobic exercise inhibits CUMS-depressed mice hippocampal inflammatory response via activating hippocampal miR-223/TLR4/MyD88-NF-κB pathway. Int J Environ Res Public Health. https://doi.org/10.3390/ijerph17082676
Obata K, Noguchi K (2004) MAPK activation in nociceptive neurons and pain hypersensitivity. Life Sci 74(21):2643–2653. https://doi.org/10.1016/j.lfs.2004.01.007
Wang D, Couture R, Hong Y (2014) Activated microglia in the spinal cord underlies diabetic neuropathic pain. Eur J Pharmacol 728:59–66. https://doi.org/10.1016/j.ejphar.2014.01.057
Montoya T, Alarcón-de-la-Lastra C, Castejón ML, Ortega-Vidal J, Altarejos J, Sánchez-Hidalgo M (2021) (-)-Methyl-oleocanthal, a new oleocanthal metabolite reduces LPS-induced inflammatory and oxidative response: molecular signaling pathways and histones epigenetic modulation. Antioxidants (Basel). https://doi.org/10.3390/antiox11010056
Svensson CI, Schäfers M, Jones TL, Powell H, Sorkin LS (2005) Spinal blockade of TNF blocks spinal nerve ligation-induced increases in spinal P-p38. Neurosci Lett 379(3):209–213. https://doi.org/10.1016/j.neulet.2004.12.064
Li J, Shi H, Liu H, Dong F, Liu Z, Lu Y, Chen L, Bao L, Zhang X (2020) Nerve injury-induced neuronal PAP-I maintains neuropathic pain by activating spinal microglia. J Neurosci 40(2):297–310. https://doi.org/10.1523/jneurosci.1414-19.2019
Svensson CI, Marsala M, Westerlund A, Calcutt NA, Campana WM, Freshwater JD, Catalano R, Feng Y, Protter AA, Scott B, Yaksh TL (2003) Activation of p38 mitogen-activated protein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J Neurochem 86(6):1534–1544. https://doi.org/10.1046/j.1471-4159.2003.01969.x
Sung CS, Wen ZH, Chang WK, Chan KH, Ho ST, Tsai SK, Chang YC, Wong CS (2005) Inhibition of p38 mitogen-activated protein kinase attenuates interleukin-1beta-induced thermal hyperalgesia and inducible nitric oxide synthase expression in the spinal cord. J Neurochem 94(3):742–752. https://doi.org/10.1111/j.1471-4159.2005.03226.x
Gao YH, Li CW, Wang JY, Tan LH, Duanmu CL, Jing XH, Chang XR, Liu JL (2017) Effect of electroacupuncture on the cervicospinal P2X7 receptor/fractalkine/CX3CR1 signaling pathway in a rat neck-incision pain model. Purinergic Signal 13(2):215–225. https://doi.org/10.1007/s11302-016-9552-1
Clark AK, Wodarski R, Guida F, Sasso O, Malcangio M (2010) Cathepsin S release from primary cultured microglia is regulated by the P2X7 receptor. Glia 58(14):1710–1726. https://doi.org/10.1002/glia.21042
Ji RR, Woolf CJ (2001) Neuronal plasticity and signal transduction in nociceptive neurons: implications for the initiation and maintenance of pathological pain. Neurobiol Dis 8(1):1–10. https://doi.org/10.1006/nbdi.2000.0360
Gong X, Jiang J, Zhang M (2016) Exercise preconditioning reduces neonatal incision surgery-induced enhanced hyperalgesia via inhibition of P38 mitogen-activated protein kinase and IL-1β, TNF-α release. Int J Dev Neurosci 52:46–54. https://doi.org/10.1016/j.ijdevneu.2016.05.008
Funding
This work was supported by the Science and Technology Department of Sichuan Province (2022NSFSC0667), and the Sports Medicine Key Laboratory of Sichuan Province (2023-A031).
Author information
Authors and Affiliations
Contributions
LK is the corresponding author of the review. MJW is the first author and is responsible for collecting materials and writing the first draft of the manuscript. XYJ, YZW, BRY and QL helped in organizing the information. XYJ, YZW and HH edited the review table and figure. All authors contributed to manuscript revision, read, and approved the submitted version.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, MJ., Jing, XY., Wang, YZ. et al. Exercise, Spinal Microglia and Neuropathic Pain: Potential Molecular Mechanisms. Neurochem Res 49, 29–37 (2024). https://doi.org/10.1007/s11064-023-04025-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-023-04025-4