
Abstract
Parkinson’s disease is a neurodegenerative disease affecting mainly the elderly population. It is characterized by the loss of dopaminergic neurons of the substantia nigra pars compacta region. Parkinson’s disease patients exhibit motor symptoms like tremors, rigidity, bradykinesia/hypokinesia, and non-motor symptoms like depression, cognitive decline, delusion, and pain. Major pathophysiological factors which contribute to neuron loss include excess/misfolded alpha-synuclein aggregates, microglial cell-mediated neuroinflammation, excitotoxicity, oxidative stress, and defective mitochondrial function. Sigma-1 receptors are molecular chaperones located at mitochondria-associated ER membrane. Their activation (by endogenous ligands or agonists) has shown neuroprotective and neurorestorative effects in various diseases. This review discusses the roles of activated Sig-1 receptors in modulating various pathophysiological features of Parkinson’s disease like alpha-synuclein aggregates, neuroinflammation, excitotoxicity, and oxidative stress.
Similar content being viewed by others

Introduction
Parkinson’s disease (PD) is one of the most common neurodegenerative diseases. The prevalence of PD is approximately 1–4% of the population over 60–80 years of age [1]. Statistics say more than ten million people have been diagnosed with PD and the numbers are still soaring [2]. The pathognomonic feature in PD post-mortem brains is Lewi bodies since these cytoplasmic inclusions are formed by the aggregation of the alpha-synuclein. Alpha-synuclein is a regulatory protein that controls many neuronal functions including neurotransmitter release [3, 4]. In PD, the gene encoding alpha-synuclein gets mutated and hence causes its aggregation. This depletes the neuronal dopamine level resulting in symptoms like tremors, rigidity, changes in gait, and bradykinesia/hypokinesia. Moreover, the pathogenic alpha-synuclein aggregate promotes neurodegeneration [3]. The post-mortem studies of the brains of PD patients show inflammation in the basal ganglia region [5, 6]. Further, overactivation of NMDA (N-methyl-d-aspartate) receptors causes excitotoicity which causes neuronal apoptosis. The exact cause of PD is still a matter of debate but various pieces of evidence have pointed to oxidative stress and genetics as etiological factors [7, 8]. The conventional pharmacological options for PD provide symptomatic relief mainly. Thus, an unmet need exists to bring new disease-modifying strategies for the treatment of PD.
The main pathophysiological factors associated with PD are excess/misfolded alpha-synuclein aggregates, microglial cell-mediated neuroinflammation, excitotoxicity, oxidative stress, and defective mitochondrial function. Disruption of mitochondrial structure and function is one of the main mechanisms in the pathogenesis of PD. Mitochondria-associated endoplasmic reticulum membrane (MAM) of neurons of the substantia nigra region have ubiquitous expression of Sigma-1 receptors (S1Rs). The S1Rs are molecular chaperones and are associated with multiple functions. S1Rs promote cell survival by activation of the defence mechanisms. S1R activation (by endogenous ligands or agonists) has shown neuroprotective and neurorestorative effects in various diseases [9,10,11].
Structure and Function of S1R
Sigma receptors are intracellular proteins expressed throughout the body including the central nervous system. Initially, sigma receptors were defined as a subclass of opioid receptors however later studies confirmed that sigma receptors are non-opioid receptors [12, 13]. Sigma receptors have two types viz. Sigma-1 and Sigma-2. The former has a prominent role in neurodegenerative diseases whereas the latter showed importance in cancer. S1Rs are intracellular chaperones with a molecular weight 29-kDa and total 223 amino acids. It does not have homology with any other mammalian protein except the yeast sterol isomerase ERG2, where it shares only 60 percent of the amino acid sequence. The amino acid sequence identity is 30.3 percent [14]. S1R resides on MAM and is neither an ionotropic receptor nor a metabotropic receptor. This 29-kDa protein has two hydrophobic domains at N-terminus and at C-terminus, it has domains for molecular chaperone activity. The centre of S1R has membrane-spanning α-helices [15,16,17,18]. The N-terminus and C-terminus lie on the same side with N-terminus located at cytosol and C-terminus located at the endoplasmic reticulum. The gene that encodes S1R i.e. SIGMAR1 is ~7 kbp long situated on chromosome 9, band p13. It has 4 exons and 3 introns with exon 3 being the shortest (93 bp) and exon 4 being the longest (1132 bp) [19,20,21]. S1Rs perform crucial roles in the cell like modulation of endoplasmic reticulum (ER) stress, mitochondrial function, glial activity, oxidative stress, and regulation of Ca+ influx. Activation of S1Rs (by endogenous ligands or agonists) decreases misfolded protein-induced ER stress, neuroinflammation, excitotoxicity, and oxidative stress. Thus, S1R poses itself as a promising target and S1R agonist, a valuable extension in pharmacological classes of anti-parkinsonian agents [22,23,24,25].
Role of S1Rs in Managing Proteome
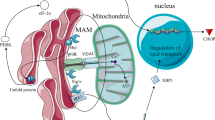
The maintenance of cellular proteins at an optimum level is of utmost importance to govern cell survival. It has been observed that excess or misfolded alpha-synuclein aggregates disturb cellular physiology and contribute to neuronal degeneration in PD [26]. Rectifying the faulty conformation or enhancing the degradation or suppressing the genesis of alpha-synuclein can be a promising strategy to tackle PD. S1Rs being molecular chaperones, control the cellular protein concentration. Excess/misfolded protein induces ER stress which activates two programs viz. unfolded protein response (UPR) and ER overload response (EOR). UPR has 3 sensor proteins (i) IRE1 (inositol-requiring enzyme 1) (ii) PERK (protein kinase RNA-activated-like endoplasmic reticulum kinase) (iii) ATF6 (activating transcription factor 6) which works to bring down ER stress [15, 22, 24, 27,28,29,30].
IRE1 undergoes dimerization and phosphorylation to get converted into its active state. Activated IRE1 splices XBP1 (X-box binding protein 1) mRNA alters the transcription of UPR genes and result in upregulation of ER chaperones. In addition, upregulation of ER chaperones occurs when ATF6 gets activated. These ER chaperones then perform ER-associated protein degradation (ERAD). The activation of PERK reduces global protein translation by inhibiting eIF-2α (eukaryotic translation initiation factor 2 α). PERK activation can also trigger apoptosis. Sustained PERK activation upregulates CHOP (C/EBP-homologous protein) which then inhibits the expression of Bcl-2 (anti-apoptotic factor) and upregulates pro-apoptotic BH3-only proteins expression. This leads to the activation of Bak- and Bax-dependent apoptosis [24]. The S1R aids in modulating the activities of IRE1, PERK, and ATF6 to a significant extent. ER stress, S1R ligand or agonist stimulation dissociates S1R from BiP (binding immunoglobulin protein) and stabilizes IRE1 (Fig. 1). S1R remains dormant when attached to BiP and performs maximum when dissociated. During ER stress, there is upregulation of S1R [15, 22, 24, 31, 32]. S1R agonism can help in alpha-synuclein clearance and prevent neurodegeneration in PD. With regards to EOR, it has been observed that its activation enhances reactive oxygen species (ROS) formation and NF-κB (nuclear factor kappa B) activation [15].

Modulation of misfolded protein-induced ER stress by sigma 1 receptor
Role of S1Rs in Modulating Neuroinflammation
Microglia are macrophage-derived immune cells of the central nervous system (CNS) and constitute 10% of the total cellular population of CNS [33]. Under sound physiological conditions, microglial cells are responsible for many functions like surveillance, neurogenesis, cognition, pathogen destruction, etc. [34]. However, in a pathological state like PD, microglial cells cause neuroinflammation and contribute to neurodegeneration. Further, neurodegeneration leads to neuroinflammation [35]. So far, two phenotypes of microglia have been discovered viz. M1 (classic) and M2 (alternative) [36]. The former is pro-inflammatory and detrimental to CNS whereas the latter is anti-inflammatory and neuroprotective. In PD, M1-type microglial activation releases nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, ROS, and pro-inflammatory cytokines (interleukin-1β,6 and tumor necrosis factor-α) which leads to neuroinflammation [37, 38]. Contrastingly, activation of M2-type microglia decreases pro-inflammatory cytokine levels and performs neuronal repair and regeneration [24, 39, 40].
S1Rs are also expressed on microglial cells and modulate microglial activities. Francardo et al. has demonstrated that S1R activation through PRE-084 and pridopidine reduces CD-68 positive microglial cells in the striatum and substantia nigra region of C57Bl/6 mice. In addition, there was a significant enhancement in TH+ cells suggesting neuroprotective properties of S1R agonists-PRE-084 and pridopidine. Further, the activity of PRE-084 and pridopidine was lost in S1R KO mice, confirming the agonist’s activity via S1R [41, 42]. Afobazole, a clinically approved anxiolytic in Russia, was reported to have affinity towards S1R. Cuevas et al. studied the effects of afobazole in ischemic stroke and found that S1R activation by afobazole decreases microglial migration and inflammatory response. For microglia to activate and migrate, the influx of cations (Ca2+, Na2+) through purinergic receptors (P2X and P2Y), which are localized on microglia, is needed. Activation of these receptors is governed by ATP (ligand of P2X and P2Y). Activated S1R is found to block these receptors and hence possess anti-inflammatory properties (Fig. 2) [33, 43]. Hall et al. reported that 1,3-Di-o-tolylguanidine (a potent S1R agonist) suppresses microglial migration, response, and morphology in a murine model of stroke. Their study suggests that S1R activation regulates various aspects of microglial function [44]. The probable mechanism could be the enhancement of anti-inflammatory cytokines i.e. interleukin-10 and 4 by S1R agonist. These anti-inflammatory cytokines inhibit inflammatory cytokines and hence prevent neuroinflammation (Fig. 2) [9, 45]. Another sigma ligand-SR-31747 was found to inhibit tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) secretion evoked by lipopolysaccharide (LPS) in rats [46]. Similar results were obtained with the study of Wu et al., but they used a different S1R ligand i.e. SKF89359 [47]. All the aforementioned data indicates the anti-inflammatory properties of S1R ligands/agonists.

Modulation of neuroinflammation by activation of sigma 1 receptor
Role of S1R in Modulating Excitotoxicity
In the brain, the balance between excitatory and inhibitory neural signals is crucial for healthy neuronal functioning and viability. The excitatory signals are mediated by Glutamate-a major excitatory neurotransmitter. The arrival of action potential/signal stimulates the synaptic vesicle to release glutamate into the synaptic cleft. The glutamate then binds to glutamatergic receptors (majorly NMDA) and transmits excitatory signals. However, in PD, there is an excess release of glutamate in the synaptic cleft which leads to overactivation and sustained activation of NMDA receptors. This overactivation promotes a multifold increase in Ca2+ influx within the cell which disturbs the intracellular environment, especially mitochondria. The mitochondria begin to malfunction and release protease which destroys mitochondria. The mitochondrial electron transport chain (ETC) gets disrupted and causes ROS generation. This phenomenon is termed as ‘excitotoxicity’ and it significantly influences the demise of neurons in PD [24, 48,49,50]. Various studies suggest a link between glutamate-mediated excitotoxicity and neuroinflammation in PD. However, there is a controversy that whether neuroinflammation is a detrimental outcome of excitotoxicity or is a cause. Studies suggested that pathological mutations cause changes in the level of glutamate receptors and transporters, implying that excitotoxicity leads to pathology. Excess of glutamate overstimulates glutamatergic receptors of microglial cells which cause neuroinflammation [48, 51]. Contrastingly, some studies suggest that microglial activation releases excessive glutamate stored within them which accelerates neurotoxicity and a strong inflammatory response is observed [52].
S1R activation attenuates glutamate-mediated excitotoxicity in numerous diseases. Selective S1R ligands or non-selective Sigma ligands have shown neuroprotective properties through modulating excitotoxicity. Shimazu et al. tested the effects of one S1R ligand and three sigma ligands on midbrain cell cultures challenged with 100 µM of NMDA. They observed a significant reduction in excitotoxicity and elevation in cell viability using the immunostaining technique. They proposed two mechanisms viz. direct mechanism and indirect mechanism by which these ligands exerted neuroprotective effects. The direct mechanism is related to the binding of sigma receptor ligands to the PCP and NR1/2B site of NMDA receptors and antagonizing them. The indirect mechanism is concerned with the interaction of S1R with the PLc (phospholipase C) pathway (Fig. 3). As the activity of the NMDA receptor is governed by protein kinase C (PKC), indirectly depleting its level may attenuate excitotoxicity. [53, 54]. Decoster et al. reported similar results using rat cortical neurons [55, 56]. Lu et al. has demonstrated that S1R agonist-SKF 10047 ([2S-(2α,6α,11R*]-1,2,3,4,5,6-hexahydro-6,11-dimethyl-3-(2-propenyl)-2,6-methano-3-benzazocin-8-ol) decreases excitotoxicity by modulation of Ca2+ influx (Fig. 3). The SKF 10047 decreases cytosolic Ca2+ levels through N-Type and P/Q-Type calcium channels and thereby decreases glutamate levels. The researchers checked the effects of PKC inhibition on glutamate-mediated excitotoxicity and found that the addition of GF109203X (PKC inhibitor) decreases the glutamate release, verifying the involvement of PKC in mediating excitotoxity [57].

Attenuation of excitotoxicity by sigma 1 receptor
Role of S1Rs in Alleviating Oxidative Stress
ROS are unstable molecules generated by various metabolic reactions that readily react with any component of the cell. They are harmless at physiological concentration and reversely, they are useful under certain cases like a microbial invasion. However, oxidative stress occurs when there is an imbalance between the production and accumulation of ROS and cells fail to combat the detrimental effects of ROS. Oxidative stress is believed to be one of the etiological factors for PD. The ROS reacts with lipids, proteins, and even DNA (deoxyribonucleic acid) which promotes neuronal death. Polyunsaturated fatty acids that are expressed abundantly on the plasma membrane are highly prone to ROS and oxidative stress disrupts the membrane permeability and fluidity. ROS reacts with alpha-synuclein and promotes its aggregation [58]. DNA damage done by ROS disturbs the coding and gene expression. The production of ROS can also be a consequence of a high level of cytosolic dopamine. Excess dopamine can readily be oxidized for the generation of ROS [8, 59,60,61].
S1Rs are reported to protect the cells against oxidative stress. Its activation attenuates ROS accumulation, upregulates endogenous antioxidants, and thereby decreases oxidative stress [24, 25]. Wang et al. reported that S1R activation by (+)-pentazocine attenuated oxidative stress and enhanced cone cell viability. The underlying mechanism proposed by the authors is the modulation of Nrf2 (nuclear factor erythroid 2-related factor). In the absence of oxidative stress, Nrf2 is bounded to KEAP1 (kelch-like ECH-associated protein 1) which enables the degradation of excess Nrf2. During oxidative stress, Nrf2 detaches from KEAP1 and translocates to the nucleus where it binds to ARE (antioxidant response element). ARE is an enhancer sequence found in the promotor region of genes expressing antioxidants [62]. The (+)-pentazocine does not interfere with Nrf2-KEAP1 binding but enhances Nrf2-ARE binding (Fig. 4). (+)-pentazocine increases the expression of Nrf2. The viability of cone cells was lost when (+)-pentazocine was administered to Nrf2 KO mice, verifying the mechanism of S1R ligand [63]. Tuerxun et al. has evaluated the effects of SA4503 (1-(3,4-dimethoxyphenethyl)-4-(3-phenylpropyl) piperazine dihydrochloride)-an S1R agonist on cultured cortical neurons and found that SA4503 inhibited the oxidative stress-induced neuronal death by downregulating MAPK/ERK (mitogen-activated protein kinase/extracellular signal-regulated kinase) pathway (Fig. 4). Cortical neurons when exposed to H2O2-induced oxidative stress lead to overactivation and sustained activation of ERK1/2. Activated ERK 1/2 controls many cellular functions including cell proliferation and cell survival but overactivated ERK1/2 contributes to oxidative stress [64]. Tuerxun et al. has demonstrated that SA4503 decreases the activation of ERK 1/2 and thereby inhibits oxidative stress. Sustained activation of ERK 1/2 leads to glutamate-mediated oxidative toxicity and so the researchers have tested the effect of SA4503 on glutamate receptors. They found that SA4503 downregulates the ionotropic glutamate receptors especially, GluR1 [65]. Tsai et al. has done a complete gene expression analysis of S1R using the Microarray technique. They showed that S1R depletion enhances ROS generation, indicating a direct correlation between S1R and ROS. Their data suggests that S1R deficiency upregulates many antioxidant genes [66].

Modulation of ROS by sigma 1 receptor
S1R as a Target for the Treatment of Parkinson’s Disease
The aforementioned sections discussed the role of S1R activation in neuroprotection. Thus, S1R agonists can be potential agents for preventing neurodegeneration. Since the discovery of S1Rs in 1976, their agonists have been tested in-vitro, preclinically and clinically (Tables 1, 2 and 3). Wang et al. has used PRE-084—a selective S1R agonist to evaluate its effects on mitophagy in MPTP induced PD model. PRE-084 enhanced neuronal mitophagy and thereby promoted the survival of neurons. The underlying mechanism was identified as S1R mediated mitophagy through PINK1/parkin pathway. The mitophagy is governed by PINK1 and activated S1R stabilizes PINK1 leading to the enhancement of mitophagy [67]. Francardo et al. used the same S1R agonist i.e. PRE-084 to study its neuroprotective properties. They used male C57BL/6 mice for the experimental study and PD was induced by stereotaxically injecting 6-hydroxydopamine (6-OHDA) into basal ganglia. Chronic treatment (5 weeks) with PRE-084 (0.3 or 1 mg/kg; s.c.) improved motor impairment in mice, evaluated by cylinder, stepping, and spontaneous rotational activity tests. There was a significant enhancement in TH+ cells in the substantia nigra region. The microglial activation was reduced and dopamine levels were elevated in the treatment group (Table 1). Studies on the impact of S1R KO on the activity of PRE-084 showed that the activity was lost in S1R KO mice [41]. In 2019, Francardo et al. again conducted the same experimental study with a different S1R agonist pridopidine [42]. Pridopidine is reported to improve motor abnormalities in an animal model of Huntington’s disease [68]. The pridopidine at doses of 0.3 or 1 mg/kg s.c. showed similar neuroprotective and neurorestorative effects as observed with PRE-084. Pridopidine enhanced the neurotrophic factors like GDNF (glial cell neurotrophic factor), BDNF (brain-derived neurotrophic factor), and ERK1/2. Similarly, the activity of pridopidine was lost in S1R KO mice [42].
Voronin et al. has demonstrated the neuroprotective effects of afobazole- an anxiolytic with S1R agonistic property. Parkinsonism was induced in ICR mice using 6-OHDA by stereotaxy. The mice treated with 2.5 mg/kg i.p. of afobazole for 2 weeks (either beginning from the 1st day of 6-OHDA injection or the 14th day of 6-OHDA injection) enhanced TH+ cells count and the latency to fall in the rotarod test. Using HPLC-ED (high-performance liquid chromatography-electrochemical detector), striatal dopamine content was measured and was found to be increased in the treatment group (Table 1). The activity of afobazole was lost when BD-1047 (Sig-1 R antagonist) was administered before afobazole administration [69, 70]. Due to these properties, the afobazole was selected for clinical trials. In 2019, Phase II trials commenced investigating its safety and effectiveness for the treatment of levodopa-induced dyskinesia in patients with PD (Clinical trial ID: NCT03922711). A total of 23 participants were recruited and the trial was double-blinded and randomized. However, due to coronavirus disease 2019 (COVID-19) pandemic, the trial was terminated and no significant data was collected.
Conclusion
In PD, neurodegeneration occurs because of numerous pathophysiological factors like pathogenic alpha-synuclein aggregation, neuroinflammation, excitotoxicity, ROS, etc. Activation of S1Rs by agonists provides neuroprotection via different mechanisms. S1R KO mice have shown increased pathogenic alpha-synuclein aggregation and S1R agonists have shown increased degradation of alpha-synuclein. This confirms the role of S1R in alpha-synuclein aggregation. In addition, S1R activation decreases microglial activation and inflammatory response. S1R agonists antagonize NMDA receptors thus inhibiting glutamate-mediated excitotoxicity. Further, S1R agonists modulate gene expression of endogenous antioxidants. All these properties make S1R an ideal therapeutic target for PD. S1R agonists have shown promising results in animal models of PD. Although the results of preclinical studies look promising, detailed clinical studies are required. A thorough investigation will aid in dragging the molecules from the bench to the bedside.
Data Availability
Not applicable.
Abbreviations
- PD:
-
Parkinson’s disease
- NMDA:
-
N-methyl-d-aspartate
- MCs:
-
Molecular chaperones
- S1Rs:
-
Sigma-1 receptors
- MAM:
-
Mitochondria-associated ER membrane
- ROS:
-
Reactive oxygen species
- ER:
-
Endoplasmic reticulum
- UPR:
-
Unfolded protein response
- EOR:
-
ER overload response
- IRE1:
-
Inositol-requiring enzyme
- PERK:
-
Protein kinase RNA-activated-like endoplasmic reticulum kinase
- ATF6:
-
Activating transcription factor 6
- XBP1:
-
X-box binding protein 1
- ERAD:
-
ER-associated protein degradation
- eIF-2α:
-
Eukaryotic translation initiation factor 2 α
- CHOP:
-
C/EBP-homologous protein
- BiP:
-
Binding immunoglobulin protein
- NF-κB:
-
Nuclear factor kappa B
- PRE-084:
-
2-Morpholin-4-ylethyl-1-phenylcyclohexane-1-carboxylate
- PINK1:
-
PTEN-induced kinase 1
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- KO:
-
Knockout
- CNS:
-
Central nervous system
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- TNF-α:
-
Tumor necrosis factor-α
- IFN-γ:
-
Interferon-γ
- LPS:
-
Lipopolysaccharide
- ETC:
-
Electron transport chain
- PLc:
-
Phospholipase C
- PKC:
-
Protein kinase C
- SKF 10047:
-
[2S-(2α,6α,11R*]-1,2,3,4,5,6-hexahydro-6,11-dimethyl-3-(2-propenyl)-2,6-methano-3-benzazocin-8-ol)
- DNA:
-
Deoxyribonucleic acid
- Nrf2:
-
Nuclear factor erythroid 2-related factor
- KEAP1:
-
Kelch-like ECH-associated protein 1
- SA4503:
-
(1-(3,4-Dimethoxyphenethyl)-4-(3-phenylpropyl) piperazine dihydrochloride)
- MAPK/ERK:
-
Mitogen-activated protein kinase/extracellular signal-regulated kinase
- 6-OHDA:
-
6-Hydroxydopamine
- GDNF:
-
Glial cell neurotrophic factor
- BDNF:
-
Brain-derived neurotrophic factor
- ERK1/2:
-
Extracellular signal-regulated kinase 1/2
- HPLC-ED:
-
High-performance liquid chromatography-electrochemical detector
- COVID-19:
-
Coronavirus disease 2019
References
de Lau LML, Breteler MMB (2006) Epidemiology of Parkinson’s disease. Lancet Neurol 5:525–535. https://doi.org/10.1016/S1474-4422(06)70471-9
Nwabufo CK, Aigbogun OP (2022) Diagnostic and therapeutic agents that target alpha-synuclein in Parkinson’s disease. J Neurol 269:5762–5786. https://doi.org/10.1007/s00415-022-11267-9
Burré J, Sharma M, Südhof TC (2018) Cell biology and pathophysiology of α-synuclein. Cold Spring Harb Perspect Med 8:a024091. https://doi.org/10.1101/cshperspect.a024091
Tomkins JE, Manzoni C (2021) Advances in protein-protein interaction network analysis for Parkinson’s disease. Neurobiol Dis 155:105395. https://doi.org/10.1016/j.nbd.2021.105395
Araújo B, Caridade-Silva R, Soares-Guedes C et al (2022) Neuroinflammation and Parkinson’s disease-from neurodegeneration to therapeutic opportunities. Cells. https://doi.org/10.3390/cells11182908
Tansey MG, Wallings RL, Houser MC et al (2022) Inflammation and immune dysfunction in Parkinson disease. Nat Rev Immunol 22:657–673. https://doi.org/10.1038/s41577-022-00684-6
Schapira AH, Jenner P (2011) Etiology and pathogenesis of Parkinson’s disease. Mov Disord 26:1049–1055. https://doi.org/10.1002/mds.23732
Jiang T, Sun Q, Chen S (2016) Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog Neurobiol 147:1–19. https://doi.org/10.1016/j.pneurobio.2016.07.005
Ajmo CTJ, Vernon DOL, Collier L et al (2006) Sigma receptor activation reduces infarct size at 24 hours after permanent middle cerebral artery occlusion in rats. Curr Neurovasc Res 3:89–98. https://doi.org/10.2174/156720206776875849
Gaja-Capdevila N, Hernández N, Navarro X, Herrando-Grabulosa M (2021) Sigma-1 receptor is a pharmacological target to promote neuroprotection in the SOD1(G93A) ALS mice. Front Pharmacol 12:780588. https://doi.org/10.3389/fphar.2021.780588
Geva M, Gershoni-Emek N, Naia L et al (2021) Neuroprotection of retinal ganglion cells by the sigma-1 receptor agonist pridopidine in models of experimental glaucoma. Sci Rep 11:21975. https://doi.org/10.1038/s41598-021-01077-w
Martin WR, Eades CG, Thompson JA et al (1976) The effects of morphine- and nalorphine- like drugs in the nondependent and morphine-dependent chronic spinal dog. J Pharmacol Exp Ther 197:517–532
Rousseaux CG, Greene SF (2016) Sigma receptors [σRs]: biology in normal and diseased states. J Recept Signal Transduct Res 36:327–388. https://doi.org/10.3109/10799893.2015.1015737
Ruoho AE, Chu UB, Ramachandran S et al (2012) The ligand binding region of the sigma-1 receptor: studies utilizing photoaffinity probes, sphingosine and N-alkylamines. Curr Pharm Des 18:920–929. https://doi.org/10.2174/138161212799436584
Hayashi T, Tsai S-Y, Mori T et al (2011) Targeting ligand-operated chaperone sigma-1 receptors in the treatment of neuropsychiatric disorders. Expert Opin Ther Targets 15:557–577. https://doi.org/10.1517/14728222.2011.560837
Sánchez-Blázquez P, Cortés-Montero E, Rodríguez-Muñoz M et al (2020) The Sigma 2 receptor promotes and the Sigma 1 receptor inhibits mu-opioid receptor-mediated antinociception. Mol Brain 13:150. https://doi.org/10.1186/s13041-020-00676-4
Ossa F, Schnell JR, Ortega-Roldan JL (2017) A review of the human sigma-1 receptor structure. Adv Exp Med Biol 964:15–29. https://doi.org/10.1007/978-3-319-50174-1_3
Ryskamp DA, Korban S, Zhemkov V et al (2019) Neuronal sigma-1 receptors: signaling functions and protective roles in neurodegenerative diseases. Front Neurosci 13:862. https://doi.org/10.3389/fnins.2019.00862
Hayashi T (2019) The sigma-1 receptor in cellular stress signaling. Front Neurosci 13:733. https://doi.org/10.3389/fnins.2019.00733
Sharma N, Patel C, Shenkman M et al (2021) The Sigma-1 receptor is an ER-localized type II membrane protein. J Biol Chem 297:101299. https://doi.org/10.1016/j.jbc.2021.101299
Prasad PD, Li HW, Fei YJ et al (1998) Exon-intron structure, analysis of promoter region, and chromosomal localization of the human type 1 sigma receptor gene. J Neurochem 70:443–451. https://doi.org/10.1046/j.1471-4159.1998.70020443.x
Weng T-Y, Tsai S-YA, Su T-P (2017) Roles of sigma-1 receptors on mitochondrial functions relevant to neurodegenerative diseases. J Biomed Sci 24:74. https://doi.org/10.1186/s12929-017-0380-6
Ruscher K, Wieloch T (2015) The involvement of the sigma-1 receptor in neurodegeneration and neurorestoration. J Pharmacol Sci 127:30–35. https://doi.org/10.1016/j.jphs.2014.11.011
Nguyen L, Lucke-Wold BP, Mookerjee SA et al (2015) Role of sigma-1 receptors in neurodegenerative diseases. J Pharmacol Sci 127:17–29. https://doi.org/10.1016/j.jphs.2014.12.005
Penke B, Fulop L, Szucs M, Frecska E (2018) The role of sigma-1 receptor, an intracellular chaperone in neurodegenerative diseases. Curr Neuropharmacol 16:97–116. https://doi.org/10.2174/1570159X15666170529104323
Tu H-Y, Yuan B-S, Hou X-O et al (2021) α-synuclein suppresses microglial autophagy and promotes neurodegeneration in a mouse model of Parkinson’s disease. Aging Cell 20:e13522. https://doi.org/10.1111/acel.13522
Zhou Z, Wang Q, Michalak M (2021) Inositol requiring enzyme (IRE), a multiplayer in sensing endoplasmic reticulum stress. Animal Cells Syst (Seoul) 25:347–357. https://doi.org/10.1080/19768354.2021.2020901
Gardner BM, Pincus D, Gotthardt K et al (2013) Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol 5:a013169. https://doi.org/10.1101/cshperspect.a013169
Adams CJ, Kopp MC, Larburu N et al (2019) Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1. Front Mol Biosci 6:11. https://doi.org/10.3389/fmolb.2019.00011
Sano R, Reed JC (2013) ER stress-induced cell death mechanisms. Biochim Biophys Acta 1833:3460–3470. https://doi.org/10.1016/j.bbamcr.2013.06.028
Schröder M, Kaufman RJ (2005) The mammalian unfolded protein response. Annu Rev Biochem 74:739–789. https://doi.org/10.1146/annurev.biochem.73.011303.074134
Mori T, Hayashi T, Hayashi E, Su T-P (2013) Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS One 8:e76941. https://doi.org/10.1371/journal.pone.0076941
Cuevas J, Rodriguez A, Behensky A, Katnik C (2011) Afobazole modulates microglial function via activation of both sigma-1 and sigma-2 receptors. J Pharmacol Exp Ther 339:161–172. https://doi.org/10.1124/jpet.111.182816
Hickman S, Izzy S, Sen P et al (2018) Microglia in neurodegeneration. Nat Neurosci 21:1359–1369. https://doi.org/10.1038/s41593-018-0242-x
Gelders G, Baekelandt V, Van der Perren A (2018) Linking neuroinflammation and neurodegeneration in Parkinson’s disease. J Immunol Res 2018:4784268. https://doi.org/10.1155/2018/4784268
Plastira I, Bernhart E, Goeritzer M et al (2016) 1-Oleyl-lysophosphatidic acid (LPA) promotes polarization of BV-2 and primary murine microglia towards an M1-like phenotype. J Neuroinflammation 13:205. https://doi.org/10.1186/s12974-016-0701-9
Pajares M, I Rojo A, Manda G et al (2020) Inflammation in Parkinson’s disease: mechanisms and therapeutic implications. Cells. https://doi.org/10.3390/cells9071687
Tang Y, Le W (2016) Differential roles of M1 and M2 MICROGLIA in neurodegenerative diseases. Mol Neurobiol 53:1181–1194. https://doi.org/10.1007/s12035-014-9070-5
Pisanu A, Lecca D, Mulas G et al (2014) Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-γ agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson’s disease. Neurobiol Dis 71:280–291. https://doi.org/10.1016/j.nbd.2014.08.011
Jia J, Cheng J, Wang C, Zhen X (2018) Sigma-1 receptor-modulated neuroinflammation in neurological diseases. Front Cell Neurosci 12:314. https://doi.org/10.3389/fncel.2018.00314
Francardo V, Bez F, Wieloch T et al (2014) Pharmacological stimulation of sigma-1 receptors has neurorestorative effects in experimental parkinsonism. Brain 137:1998–2014. https://doi.org/10.1093/brain/awu107
Francardo V, Geva M, Bez F et al (2019) Pridopidine induces functional neurorestoration via the sigma-1 receptor in a mouse model of Parkinson’s disease. Neurotherapeutics 16:465–479. https://doi.org/10.1007/s13311-018-00699-9
Puchałowicz K, Tarnowski M, Baranowska-Bosiacka I et al (2014) P2X and P2Y receptors—role in the pathophysiology of the nervous system. Int J Mol Sci 15:23672–23704. https://doi.org/10.3390/ijms151223672
Hall AA, Herrera Y, Ajmo CTJ et al (2009) Sigma receptors suppress multiple aspects of microglial activation. Glia 57:744–754. https://doi.org/10.1002/glia.20802
Allahtavakoli M, Jarrott B (2011) Sigma-1 receptor ligand PRE-084 reduced infarct volume, neurological deficits, pro-inflammatory cytokines and enhanced anti-inflammatory cytokines after embolic stroke in rats. Brain Res Bull 85:219–224. https://doi.org/10.1016/j.brainresbull.2011.03.019
Bourrié B, Bribes E, De Nys N et al (2002) SSR125329A, a high affinity sigma receptor ligand with potent anti-inflammatory properties. Eur J Pharmacol 456:123–131. https://doi.org/10.1016/s0014-2999(02)02646-8
Wu Z, Li L, Zheng L-T et al (2015) Allosteric modulation of sigma-1 receptors by SKF83959 inhibits microglia-mediated inflammation. J Neurochem 134:904–914. https://doi.org/10.1111/jnc.13182
Iovino L, Tremblay ME, Civiero L (2020) Glutamate-induced excitotoxicity in Parkinson’s disease: the role of glial cells. J Pharmacol Sci 144:151–164. https://doi.org/10.1016/j.jphs.2020.07.011
Nguyen L, Lucke-Wold BP, Mookerjee S et al (2017) Sigma-1 receptors and neurodegenerative diseases: towards a hypothesis of sigma-1 receptors as amplifiers of neurodegeneration and neuroprotection. Adv Exp Med Biol 964:133–152. https://doi.org/10.1007/978-3-319-50174-1_10
Nguyen L, Kaushal N, Robson MJ, Matsumoto RR (2014) Sigma receptors as potential therapeutic targets for neuroprotection. Eur J Pharmacol 743:42–47. https://doi.org/10.1016/j.ejphar.2014.09.022
Zhang Y, Meng X, Jiao Z et al (2020) Generation of a novel mouse model of Parkinson’s Disease via targeted knockdown of glutamate transporter GLT-1 in the substantia nigra. ACS Chem Neurosci 11:406–417. https://doi.org/10.1021/acschemneuro.9b00609
Wang J, Wang F, Mai D, Qu S (2020) Molecular mechanisms of glutamate toxicity in Parkinson’s disease. Front Neurosci 14:585584. https://doi.org/10.3389/fnins.2020.585584
Shimazu S, Katsuki H, Takenaka C et al (2000) sigma receptor ligands attenuate N-methyl-d-aspartate cytotoxicity in dopaminergic neurons of mesencephalic slice cultures. Eur J Pharmacol 388:139–146. https://doi.org/10.1016/s0014-2999(99)00852-3
Whittemore ER, Ilyin VI, Woodward RM (1997) Antagonism of N-methyl-d-aspartate receptors by sigma site ligands: potency, subtype-selectivity and mechanisms of inhibition. J Pharmacol Exp Ther 282:326–338
DeCoster MA, Klette KL, Knight ES, Tortella FC (1995) Sigma receptor-mediated neuroprotection against glutamate toxicity in primary rat neuronal cultures. Brain Res 671:45–53. https://doi.org/10.1016/0006-8993(94)01294-r
Walker JM, Bowen WD, Walker FO et al (1990) Sigma receptors: biology and function. Pharmacol Rev 42:355–402
Lu C-W, Lin T-Y, Wang C-C, Wang S-J (2012) σ-1 Receptor agonist SKF10047 inhibits glutamate release in rat cerebral cortex nerve endings. J Pharmacol Exp Ther 341:532–542. https://doi.org/10.1124/jpet.111.191189
Dias V, Junn E, Mouradian MM (2013) The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis 3:461–491. https://doi.org/10.3233/JPD-130230
Sanders LH, Timothy Greenamyre J (2013) Oxidative damage to macromolecules in human Parkinson disease and the rotenone model. Free Radic Biol Med 62:111–120. https://doi.org/10.1016/j.freeradbiomed.2013.01.003
Song Q, Peng S, Zhu X (2021) Baicalein protects against MPP(+)/MPTP-induced neurotoxicity by ameliorating oxidative stress in SH-SY5Y cells and mouse model of Parkinson’s disease. Neurotoxicology 87:188–194. https://doi.org/10.1016/j.neuro.2021.10.003
Aborode AT, Pustake M, Awuah WA et al (2022) Targeting oxidative stress mechanisms to treat Alzheimer’s and Parkinson’s disease: a critical review. Oxid Med Cell Longev 2022:7934442. https://doi.org/10.1155/2022/7934442
Vomhof-Dekrey EE, Picklo MJS (2012) The Nrf2-antioxidant response element pathway: a target for regulating energy metabolism. J Nutr Biochem 23:1201–1206. https://doi.org/10.1016/j.jnutbio.2012.03.005
Wang J, Zhao J, Cui X et al (2019) The molecular chaperone sigma 1 receptor mediates rescue of retinal cone photoreceptor cells via modulation of NRF2. Free Radic Biol Med 134:604–616. https://doi.org/10.1016/j.freeradbiomed.2019.02.001
Guo Y-J, Pan W-W, Liu S-B et al (2020) ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med 19:1997–2007. https://doi.org/10.3892/etm.2020.8454
Tuerxun T, Numakawa T, Adachi N et al (2010) SA4503, a sigma-1 receptor agonist, prevents cultured cortical neurons from oxidative stress-induced cell death via suppression of MAPK pathway activation and glutamate receptor expression. Neurosci Lett 469:303–308. https://doi.org/10.1016/j.neulet.2009.12.013
Tsai S-Y, Rothman RK, Su T-P (2012) Insights into the Sigma-1 receptor chaperone’s cellular functions: a microarray report. Synapse 66:42–51. https://doi.org/10.1002/syn.20984
Wang M, Wan C, He T et al (2021) Sigma-1 receptor regulates mitophagy in dopaminergic neurons and contributes to dopaminergic protection. Neuropharmacology 196:108360. https://doi.org/10.1016/j.neuropharm.2020.108360
Geva M, Kusko R, Soares H et al (2016) Pridopidine activates neuroprotective pathways impaired in huntington disease. Hum Mol Genet 25:3975–3987. https://doi.org/10.1093/hmg/ddw238
Voronin MV, Kadnikov IA, Voronkov DN, Seredenin SB (2019) Chaperone sigma1r mediates the neuroprotective action of afobazole in the 6-OHDA model of Parkinson’s disease. Sci Rep 9:17020. https://doi.org/10.1038/s41598-019-53413-w
Kadnikov IA, Verbovaya ER, Voronkov DN et al (2020) Deferred administration of afobazole induces sigma1r-dependent restoration of striatal dopamine content in a mouse model of Parkinson’s disease. Int J Mol Sci. https://doi.org/10.3390/ijms21207620
Keller M, Griesmaier E, Auer M et al (2008) Dextromethorphan is protective against sensitized N-methyl-d-aspartate receptor-mediated excitotoxic brain damage in the developing mouse brain. Eur J Neurosci 27:874–883. https://doi.org/10.1111/j.1460-9568.2008.06062.x
Goguadze N, Zhuravliova E, Morin D et al (2019) Sigma-1 receptor agonists induce oxidative stress in mitochondria and enhance complex I activity in physiological condition but protect against pathological oxidative stress. Neurotox Res 35:1–18. https://doi.org/10.1007/s12640-017-9838-2
Urfer R, Moebius HJ, Skoloudik D et al (2014) Phase II trial of the sigma-1 receptor agonist cutamesine (SA4503) for recovery enhancement after acute ischemic stroke. Stroke 45:3304–3310. https://doi.org/10.1161/STROKEAHA.114.005835
Shenkman M, Geva M, Gershoni-Emek N et al (2021) Pridopidine reduces mutant huntingtin-induced endoplasmic reticulum stress by modulation of the sigma-1 receptor. J Neurochem 158:467–481. https://doi.org/10.1111/jnc.15366
Funding
None.
Author information
Authors and Affiliations
Contributions
TS: Conceptualization, Writing—Original draft preparation, LKB: Conceptualization, Reviewing and Editing.
Corresponding author
Ethics declarations
Competing Interest
The authors declare no competing interests.
Ethical Approval
Not applicable.
Consent for Publication
Not applicable.
Consent to Participate
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Siddiqui, T., Bhatt, L.K. Targeting Sigma-1 Receptor: A Promising Strategy in the Treatment of Parkinson’s Disease. Neurochem Res 48, 2925–2935 (2023). https://doi.org/10.1007/s11064-023-03960-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-023-03960-6