
Abstract
Despite similar morphological aspects, anaplastic oligodendroglial tumors (AOTs) form a heterogeneous clinical subgroup of gliomas. The chromosome arms 1p/19q codeletion has been shown to be a relevant biomarker in AOTs and to be perfectly exclusive from EGFR amplification in gliomas. To identify new genomic regions associated with prognosis, 60 AOTs from the EORTC trial 26951 were analyzed retrospectively using BAC-array-based comparative genomic hybridization. The data were processed using a binary tree method. Thirty-three BACs with prognostic value were identified distinguishing four genomic subgroups of AOTs with different prognosis (p < 0.0001). Type I tumors (25%) were characterized by: (1) an EGFR amplification, (2) a poor prognosis, (3) a higher rate of necrosis, and (4) an older age of patients. Type II tumors (21.7%) had: (1) loss of prognostic BACs located on 1p tightly associated with 19q deletion, (2) a longer survival, (3) an oligodendroglioma phenotype, and (4) a frontal location in brain. Type III AOTs (11.7%) exhibited: (1) a deletion of prognostic BACs located on 21q, and (2) a short survival. Finally, type IV tumors (41.7%) had different genomic patterns and prognosis than type I, II and III AOTs. Multivariate analysis showed that genomic type provides additional prognostic data to clinical, imaging and pathological features. Similar results were obtained in the cohort of 45 centrally reviewed–validated cases of AOTs. Whole genome analysis appears useful to screen the numerous genomic abnormalities observed in AOTs and to propose new biomarkers particularly in the non-1p/19q codeleted AOTs.
Similar content being viewed by others

Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.Avoid common mistakes on your manuscript.
Introduction
Diffuse gliomas are the most frequent primary malignant cerebral tumors in adults. They are classified by the World Health Organization (WHO) according to their phenotype (astrocytic, oligodendrocytic and oligoastrocytic) and their grade of malignancy (from WHO grades II to IV) [1, 2].
Anaplastic or WHO grade III oligodendroglial tumors (AOTs), including anaplastic oligodendrogliomas and anaplastic oligoastrocytomas, are particularly disconcerting tumors: despite similar clinical, radiological and pathological features, these tumors exhibit dramatically heterogeneous outcomes. The overall survival (OS) of AOT patients varies from 2 to 10 years [3–6]. Moreover, the diagnosis of AOTs suffers from a significant interobserver variation that complicates the apprehension of these tumors [7, 8].
Since the princeps studies conducted by Reifenberger et al. and Cairncross et al., several independent teams have validated that the chromosome arms 1p/19q codeletion is frequent in AOTs and is a relevant genomic biomarker associated with longer survival and better chemosensitivity in AOTs [4, 5, 9–12]. Interestingly, this cytogenetic imbalance has recently been linked to a chromosome translocation (1;19) (q10;p10) and has been shown to be mutually exclusive from high level gene amplification [13–15]. Classically, in the subgroup of high grade gliomas, EGFR high level amplification is strongly associated with WHO grade IV astrocytomas (glioblastomas or GBMs) [16–19]. However, it is also observed in AOTs, and its detection does not permit the conversion of a diagnosis of an AOT into that of a GBM according to strictly morphology-based WHO criteria [15, 20–23].
Taken together, these data suggest that the 1p/19q codeleted AOTs form a very singular clinico-genetic subgroup of AOTs: (1) distinct from their morphological non-1p/19q codeleted counterparts in terms of chemosensitivity and prognosis, and (2) genetically separated from EGFR-amplified AOTs because these alterations are mutually exclusive. The presence and the clinical significance of EGFR amplification in AOTs are not definitively elucidated but have been shown to be associated with poor prognosis [21, 24].
Finally, the non-1p/19q codeleted and non-EGFR-amplified AOTs remain a heterogeneous group of tumors without specific recurrent genomic background. This prompted us to conduct the present study using Bacterial Artificial Chromosome (BAC)-array-based comparative genomic hybridization (aCGH) analyzing the genomic patterns of a series of 60 AOTs included in the EORTC trial 26951. The EORTC clinical trial 26951 was a prospective randomized phase III study comparing radiotherapy plus chemotherapy with procarbazine, lomustine and vincristine (PCV) versus radiotherapy alone as first line treatment in a cohort of 368 patients with AOTs [4]. This homogeneously-treated patient population allows the determination of the significance of the various genetic profiles of AOTs.
Materials and methods
Materials
Eligible for this study were all patients that were enrolled in EORTC study 26951 and from whom frozen tumor material was available for further research. Freezing tumor samples was not mandatory in the design of the EORTC clinical trial 26951. However, high quality DNA extracted from frozen tissue was required for running aCGH. Therefore, European centers participating to the clinical trial and known to freeze tumor tissue were contacted. Eligible for this clinical trial were patients with anaplastic oligodendroglioma or oligoastrocytoma according to the WHO 1994 definition as diagnosed by the local pathologist, with at least 3 out of 5 anaplastic characteristics (nuclear polymorphism, mitosis, high cellularity, endothelial proliferation and necrosis). The details of the study are described elsewhere [4]. For the present analysis, the pathological diagnosis of the local and the central pathologists were used. For the conduct of the study and the present translational research study, approval was obtained from national and local institutional review boards according to national and local regulations; written informed consent was obtained from all patients prior to study entry.
BAC-array comparative genomic hybridization
Tumor DNA was successively extracted from frozen tumor tissue using a commercial kit (QIAamp DNA mini Kit; Qiagen, Courtaboeuf, France), digested with DpnII (Ozyme, Saint Quentin en Yvelines, France), column-purified (Qiaquick PCR purification kit; Qiagen) and labeled by the random priming technique (Bioprime DNA labelling system; Invitrogen, Cergy-Pontoise, France) with cyanine-5 (Perkin-Elmer, Wellesley, MA, USA). The same procedure was applied to control DNA with cyanine-3. Probes were cohybridized on the aCGH after the following steps: (1) ethanol-coprecipitation with 140 μg of Human Cot-1 DNA (Invitrogen), (2) resuspension in hybridization buffer containing 50% of formamide, (3) heat-denaturation at 95°C during 10 min, and (4) prehybridization at 37°C for 90 min. Thirty-seven tumors were run on the aCGH (Integragen, Paris, France) analyzing 4,500 sequence-validated BACs and 13 tumors, part of a previous study, were run on the previous version of the chip containing 3,342 BACs [15]. The 4,500 and the 3,342 BACs were spotted on the array in quadruplicate and in triplicate, respectively. Five hundred were cancer-related genes and the other BACs were picked approximately across the genome of each megabase. Arrays were washed and scanned using a 4000B scanner (Axon, Union City, CA, USA). Images were analyzed with the Genepix6.0 software (AxonA). Data were processed through the CAPweb platform to obtain Cy5/Cy3 fluorescence ratios and status (gained, lost, amplified or normal) for each BAC [25].
Fluorescent in situ hybridization (FISH) analysis
FISH experiments were conducted as previously reported [26]. Briefly, probes to 1p36, centromere 1, 19p and 19q13 were D1S32, CEP1/pUC1.77, equivalent amounts of human BACs RPCI-11 959O6/957I1/153P24 (BacPac Resources, Oakland, CA, USA) and RPCI-11 426G3 (Research Genetics, Huntsville, AL, USA) respectively. CEP1/pUC1.77, RPCI-11 959O6/957I1/153P24 were labeled with digoxigenin-16-dUTP (Roche Diagnostics, Mannheim, Germany), D1S32 with biotin-16-dUTP (Roche Diagnostics, Mannheim, Germany) and RPCI-11 426G3 with spectrum orange-dUTP (Vysis, Downers Grove, IL, USA). Paired probes were hybridized upon the pre-processed tumor section and detected using anti-digoxigenin conjugated with fluorescein isothiocyanate (FITC; Roche Diagnostics) or CY3-conjugated avidin (Brunschwig Chemie, Amsterdam, Netherlands) antibodies. Nuclei were counterstained using DAPI mixed with an antifade solution (Vector Laboratories, Burlingam, CA, USA).
Statistical analysis
From the 3,301 BACs common to the two versions of the aCGH, the BACs missing for more than 40 values (out of 60) were excluded. From the 3,115 remaining BACs, the 2,983 BACs located on the 22 autosomes were analyzed.
For the survival analysis, we considered a binary-tree method because already known data can be incorporated into the partition procedure, particularly the mutual exclusion of chromosome arms 1p/19q codeletion, with EGFR high level amplification distinguishing two singular clear-cut genomic groups.
The binary-tree procedure selects cut points for predictors that best optimize the classical Cox proportional-hazards regression score statistic. This latter statistic measures the relationship between BAC status (deleted, normal, gained) and time to event under the null hypothesis. Here, the candidate predictors (for each autosome short/long arms) were built from survival-related BACs in a classical linear combination fashion. For taking into account multiple-testing problem (numerous BACs, numerous combination of BACs, numerous tumors, numerous additional variables), we controlled the local false discovery rate (probability of having no association given the observed statistic) at a 15% threshold. [27] As mentioned above, two special candidate predictors were constructed for EGFR high level amplification and chromosome arms 1p/19q codeletion. In practice, our tree was built according to the following procedure:
-
1.
From a univariate Cox proportional-hazards regression model, we identified BACs significantly associated with the OS. From the significant BACs on chromosome 7 (containing EGFR locus), we calculated a score X which is a candidate predictor for the chromosome 7 such that:
$$ X_{j} = {\frac{{\sum\nolimits_{i = 1}^{60} {x_{ij} \times 1_{lfdr(i) < 0,15} \times 1_{chr(i) = 7} } }}{{\sum\nolimits_{i = 1}^{60} {1_{lfdr(i) < 0,15} \times 1_{chr(i) = 7} } }}} $$where x ij is the status for the BAC i and for the patient j, and lfdr(i) and and chr(i) are the estimated lfdr and the chromosome number for the BAC i, respectively.
-
2.
Adjusting the Cox proportional-hazards regression model on X, we repeated the analysis excluding BACs located on chromosome 7. From the significant BACs on chromosome arms 1p and 19q, we calculated a score Y giving the patient status according to 1p/19q such that:
$$ Y_{j} = {\frac{{\sum\nolimits_{i = 1}^{60} {x_{ij} \times 1_{lfdr(i) < 0,15} \times 1_{{chr(i) \in^{\prime}1;19}} } }}{{\sum\nolimits_{i = 1}^{60} {1_{lfdr(i) < 0,15} \times 1_{{chr(i) \in^{\prime}1;19}} } }}}. $$ -
3.
Adjusting the Cox proportional-hazards regression model on both X and Y, we repeated the analysis excluding BACs situated on chromosome 1 and 19. From the new identified region on chromosome 21 (independent from EGFR and 1p/19q), we calculated candidate predictor Z such that:
$$ Z_{j} = {\frac{{\sum\nolimits_{i = 1}^{60} {x_{ij} \times 1_{lfdr(i) < 0,15} \times 1_{chr(i) = 21} } }}{{\sum\nolimits_{i = 1}^{60} {1_{lfdr(i) < 0,15} \times 1_{chr(i) = 21} } }}} $$
A classification rule was then defined according to the three scores X, Y and Z. The cutoff values were determined so that the classification rule gives a good balance between a sufficient number of patients in each group and a good separation for the four groups. The survival curves obtained from the Kaplan–Meier method were compared using log-rank tests.
Finally, significant prognostic factors in univariate analyze were included as candidate variables in a multivariate Cox proportional-hazards regression model analysis.
Association of genetic-prognostic patterns or groups (revealed by the binary-tree method) with clinical, radiological and pathological features of tumors was evaluated using the χ 2 test. Continuous ages were compared using a Mann–Whitney test. Agreement between aCGH and FISH results were evaluated using Cohen’s kappa test. Statistical tests were performed using XLSTAT® software. A p value < 0.05 was considered as significant.
Results
Patients and tumors characteristics
Frozen material was available for 60 patients (from 4 different centers) out of the 368 patients enrolled in the EORTC trial 26951. They were 34 men and 26 women (sex ratio = 1.3). The median age of the patients at diagnosis was 48.0 years (range 19.2–67.6) WHO performance status was 0, 1 and 2 in 21, 25 and 14 cases respectively. Forty-three tumors (71.7%) were diagnosed as anaplastic oligodendroglioma and 17 (29.3%) as anaplastic oligoastrocytomas. The tumor location was frontal in 20 (33.3%) cases and elsewhere in brain in 40 cases (66.7%). Radiological contrast enhancement was seen in 34 cases (85.0%). In 7 cases (11.7%), a previous resection of a low grade tumor had been performed. With respect to treatments, a biopsy alone, a partial resection and a complete resection were performed in 2 (3.3%), 34 (56.7%) and 24 (40.0%) patients, respectively. After surgery, 29 patients (48.3%) were randomized to the “radiotherapy plus chemotherapy by PCV” arm, and 31 patients (51.7%) to the “radiation therapy alone” arm.
The characteristics of the patients and the tumors included in the present study do not differ significantly from those ones of the entire cohort of patients and tumors included in the EORTC trial 26951, except for the surgical procedure. Indeed, in the current cohort, 97% of patients were treated with surgical resection (total and partial) versus 86% in the entire cohort patients of the clinical trial (p = 0.03) (Supplementary Table 1). Similarly, among the patients and the tumors, with a centrally reviewed–validated diagnosis of AOTs, those included in the present study were very similar to those not included in the present study except for the brain location. Indeed, in the present study, 29% of the centrally reviewed–validated AOTs were located in the frontal lobes versus 48% in the cohort of tumors not included in the present study (p = 0.03) (Supplementary Table 2).
Genomic abnormalities
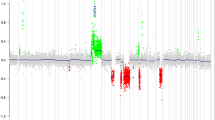
Several recurrent genomic imbalances were observed in the series. At a chromosome level, the most frequent chromosome arm gain involved chromosome arms 7q, 7p, 10p, 10q, 19p in 51.7, 40.0, 28.3, 23.3 and 21.7% of cases, respectively. The most frequently lost chromosome arms were 10q, 10p, 9p, 19q, 22q, 1p, 14q and 13q in 50.0, 41.7, 38.3, 38.3, 31.7, 30.0, 23.3 and 21.7% of tumors, respectively. At a BAC level, the most frequently gained and lost BACs were located on chromosomes 7 and 10, respectively (Fig. 1; Supplementary Tables 3 and 4). Interestingly, several BACs containing genes of interest in gliomas such as MGMT, CDKN2A, PTEN, EGFR and MET have been found abnormal in more than 50.0% of tumors (Supplementary Tables 3, 4).

Frequencies of genomic abnormalities in the entire population of anaplastic oligodendroglial tumors. The x-axis indicates the BACs and chromosome arms along the genome while the y-axis indicates the frequency of genomic alterations with genomic losses on the bottom (gray histograms) and genomic gains on top (black histograms)
FISH experiments testing chromosome arms 1p/19q and EGFR status were conducted in 53/60 and 35/60 samples, respectively. The agreement between FISH and aCGH for detection of 1p/19q codeletion and EGFR high level amplification was good (kappa = 0.6 and 0.8, respectively) (Supplementary Table 5).
BACs with prognostic value highlight four clinico-genomic groups of anaplastic oligodendroglial tumors (Figs. 2, 3, and 4)
Based on the analysis of the 2,983 BACs, the binary-tree method identified 33/2,983 relevant BACs with prognostic value located on chromosomes 1, 7 and 21. These 33 BACs with prognostic value permit the building of four clinico-genomic subgroups (Types I to IV) of AOTs (Fig. 2; Supplementary Table 6). Chromosome locations of these 33 BACs with prognostic value are reported in Fig. 3.

Genetic-prognostic tree built from the 33 relevant genetic-prognostic BACs

Chromosome location of the 33 BACs with prognostic value identified as relevant to build the genetic-prognostic tree (see Supplementary Table 4 for the full-length name and genomic position of BACs reported in this figure). a, b and c indicate chromosomes 1, 7 and 21, respectively. Along the vertical axis, each dot represents a BAC. The horizontal axis indicates chromosome distance from the telomere (d) of BACs with prognostic value identified in each chromosome. Broken arrow indicates chromosome centromere

Overall survival of anaplastic oligodendroglial tumors (AOT) patients according to the genetic-prognostic type of their tumor based on the initial diagnosis of AOT (a) and the centrally reviewed-validated diagnosis of AOT (b). Kaplan–Meier curves with x-axis and y-axis indicating the overall survival in years and the percentage of survivors, respectively. Thick continuous line, thin continuous line, thick broken line and thin broken line indicate type II, type IV, type III and type I tumors patients, respectively
Analysis based on both initial and centrally reviewed–validated pathological diagnosis
Analysis was run both on initial diagnosis and on centrally reviewed–validated pathological diagnosis. In the series of the 60 AOTs at initial diagnosis, the clinico-pathological characteristics of the entire cohort of patients and the four genetic-prognostic subgroups are in Supplementary Table 7. Interestingly, the univariate and multivariate analysis showed that genomic group provides additional prognostic information to the other variables, particularly age (Fig. 4a; Supplementary Table 8).
Among the 60 AOTs, 45 were confirmed as AOTs (31 anaplastic oligodendrogliomas and 14 anaplastic oligoastrocytomas) by the central pathological reviewer [8]. The excluded samples after central review were high grade astrocytomas (62%) and low grade gliomas (38%). Among the 45 tumors, the genomic types I, II, III and IV were observed in 15, 9, 4 and 17 tumors, respectively. Clinical, radiological and pathological characteristics of the reviewed AOTs are reported in the Table 1.
Type I AOTs, with EGFR high level amplification, were observed mainly in the elderly patients. All the type I tumors presented high cellularity, cytonuclear atypia and endothelial abnormalities. In addition, type I AOTs exhibited the higher rate of necrosis and were rarely observed in the frontal brain lobes.
Type II tumors, with loss of the BACs with prognostic value located on chromosome arm 1p (strongly associated with chromosome arm 19q loss), were exclusively oligodendrogliomas with mitosis and endothelial abnormalities. The majority of type II tumors did not present any necrosis and were located in the frontal regions.
Type III AOTs included a very small number of tumors (n = 4). Type III AOTs patients were the youngest compared to the patients from the other genomic type tumors. None of the tumors were located in the frontal lobes and only 2 of 4 tumors were contrast-enhanced on radiological examination. The lowest rates of mitosis and endothelial abnormalities were observed in this genomic subgroup.
Type IV tumors did not present either EGFR amplification or loss of the BACs with prognostic value located on chromosome arms 1p and 21q.
In the population of centrally confirmed AOTs, OS remained significantly different between the four genomic groups (p < 0.001) (Fig. 4b). Age is also a strong prognostic factor in the series of centrally reviewed–validated AOTs. Patients with genomic type I tumor were significantly older than patients with another genomic type tumor. Multivariate analysis, including age and genomic type (type I vs others or type I&III vs others), showed in both situations (type I vs others or type I&III vs others) that age and genomic type provide independent prognostic data (p < 0.0001). Therefore, difference in outcome, between genomic types, is not exclusively attributable to the age difference (Table 2).
Discussion
The diagnosis, the treatment and the prognosis prediction of AOTs remain a clinical challenge particularly in the non-1p/19q codeleted tumors [4, 5, 10]. In this latter heterogeneous group, new specific predictors of outcome and sensitivity to treatments are needed.
In a previous study on a series of gliomas of various histologies, we have shown that the 1p/19q codeletion is perfectly mutually exclusive from EGFR high level amplification [15]. These two genomic abnormalities allow identification of two clear-cut homogeneous subgroups of gliomas defined positively (EGFR-amplified vs 1p/19q codeleted tumors). These molecular abnormalities observed in 46.7% of the AOTs in the present series were used to build the first branches of the genetic-prognostic tree. Then, the statistical algorithm searched for additional biomarkers associated with OS in the non-1p/19q codeleted and non-EGFR-amplified AOTs. Since this is a post-hoc exploratory study, this needs confirmation in a second independent dataset.
In agreement with previous reports, the 1p/19q codeleted AOTs (type II) were anaplastic oligodendrogliomas located in the frontal lobes and with long OS (median OS not reached at 8 years) [4, 5, 10, 11, 28]. The present study also confirmed the interest in testing the extent of the 1p36 deletion in gliomas to distinguish the whole 1p arm loss versus focal 1p36 loss and the input of whole genomic approaches [29–31].
AOTs with EGFR amplification (type I) exhibited a higher rate of necrosis and were observed in older patients with a short OS (median ~1 year). These tumors share very similar clinico-pathological features with GBMs (“GBM-like AOT”). However, the WHO classification does not include genomic data in gliomas diagnosis. The clinical implications of EGFR amplification are not perfectly known in AOTs, but this molecular abnormality seems to be associated with unfavorable outcome [21, 24]. Similarly, the EGFR amplification prevalence in AOTs is debatable, varying from less than 10 to ~40 probably due to differences in interpretation of histological criteria by pathologists [17, 20, 23, 24, 32, 33]. In the present series, EGFR high level amplification was seen in 25% of tumors which is in agreement with the results obtained in a larger set of patients taken from the clinical trial EORTC 26951 [4, 34]. Apart from the evaluation of the 1p/19q status in AOT, assessing the EGFR gene status in AOTs may help in understanding the clinical outcome of these tumors, since they may actually be better off with treatment as is given to GBM.
Fifty-three percent of tumors showed neither 1p/19q codeletion nor EGFR high level amplification. Interestingly, the binary tree method strategy used in the present study identified chromosome 21q loss as an additional biomarker in this group of AOTs. It is observed in ~12% of cases and associated with poor prognosis (median OS = 1.6 years). To our knowledge, chromosome 21q loss has never been reported as a genomic biomarker in AOTs. In the lost BACs on chromosome 21 associated with prognosis, OLIG2 is listed. OLIG2 is a transcription factor expressed in oligodendroglial and at a lower level in astrocytic gliomas [35, 36]. This leads us to speculate that AOTs with a genomic loss of OLIG2 locus might have a lower expression of OLIG2 and thus are closer to astrocytic tumor whose prognosis is worse than their same grade oligodendroglial counterpart [3]. Type IV AOTs is defined negatively by the absence of the genomic abnormalities seen in the other types of AOTs and includes a significant number of tumors (42%). This highlights the complexity and the heterogeneity of AOTs and supports additional correlative studies including clinical, pathological and molecular characteristics of patients and tumors in this tumor group searching for other key markers.
Multivariate analysis revealed that genomic information provides putatively interesting additional prognostic information to clinical, radiological and pathological data. Indeed, the two independent prognostic factors identified in the present series were age and genomic pattern. In contrast, tumor phenotype, necrosis and contrast enhancement were not independent prognostic factors in the present series. Multivariate analysis was impossible in the series of 45 AOTs confirmed at review due to the small number of tumors and the numerous prognostic factors identified in the univariate analysis. In a larger series of patients from the same trial, using FISH on paraffin-embedded tissue samples, 1p/19q codeletion, phenotype and necrosis but not EGFR amplification were found to be of prognostic value [34]. The prognostic significance of these markers was also checked in the series of 45 AOTs reviewed. The deletion of the BACs located on chromosome arm 1p (tightly associated with the deletion of the chromosome arm 19q) had also prognostic impact in the series of 45 AOTs reviewed (p = 0.0042). However, EGFR amplification did have prognostic value (p < 0.0001) in this series, while neither the presence of tumor necrosis nor the tumor phenotype were associated with OS. These differences might be explained by several reasons. The present series is much smaller and therefore has limited power. Despite the good agreement between aCGH and FISH techniques, we have noticed a few discrepancies putatively due to interpretation issues, technical limits and/or genomic tumor heterogeneity. These disagreements may explain some of the differences observed between the two studies. Finally, necrosis and the diagnosis of an oligoastrocytic tumor are tightly associated with EGFR high level amplification and could summarize prognostic information provided by EGFR high amplification in our study.
To our knowledge, the present study is the first dedicated to clinico-genomic correlations specifically in the group of AOTs using BAC-array CGH and a binary tree approach. It is noteworthy that the present and the entire population of the EORTC 26951 trial are very similar in terms of 1p/19q status and pathology.
Several studies conducted in gliomas have shown the feasibility and the interest of array CGH for identification of relevant copy number abnormalities and candidate biomarkers [15, 22, 30, 37–45].
Kitange et al. and Trost et al. have identified the chromosome 8q gain as a biomarker of worse prognosis in series of 35 oligodendroglial tumors and 49 anaplastic oligodendrogliomas, respectively [46, 47]. The prevalence of chromosome 8q gain in the present series (23%) is similar to the one reported by Kitange et al. However, we did not see any prognostic value of chromosome 8q in our series (p = 0.2) which included exclusively WHO grade III tumors in contrast to Kitange’s series which included both grade II and III tumors. In an independent set of anaplastic oligodendroglioma,s we recently showed that high level gene amplification, whatever the gene involved, is associated with a poor prognosis [48].
More recently, Giannini et al. have reported a translational study investigating prognostic factors, including the chromosome arms 1p/19q status assessed by FISH, in AOTs from the Trial 9402 conducted by the Radiation Therapy Oncology Group [6]. Interestingly, they showed that pathological and molecular are synergistic, providing complementary prognosis information. Indeed, the histo-molecular signature “1p/19q codeleted plus classic morphology” was reported to the best predictor of better outcome in AOTs [6].
To conclude, AOTs are still a very heterogeneous group of tumors with different outcome and genomic background. The 1p/19q codeletion is a favorable prognostic marker and is currently used as an eligibility criterion for trials on anaplastic gliomas (EORTC 26053). Based on our results and other studies, there is growing evidence that additional prognostic markers are emerging particularly in the non-1p/19q codeleted AOTs, such as necrosis, oligoastrocytoma phenotype, classic oligodendroglioma morphology, gene amplification, 8q gain and 21q loss although they need additional validating studies in independent datasets. New generations of genomic arrays, single nucleotide polymorphisms-based or oligonucleotides-based, more resolutive than aCGH and feasible, at least for some of them, with different kinds of tissue (i.e. frozen or fixed), have been implemented quite recently in different laboratories. These tools will be extremely important to dissect in-depth genomic profiles of AOTs and, through clinico-molecular correlations, to identify new biomarkers in this group of glial tumors [49–51].
References
Kleihues P, Louis DN, Scheithauer BW et al (2002) The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol 61:215–225
Louis DN, Ohgaki H, Wiestler OD et al (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109
Louis DN, Holland EC, Cairncross JG (2001) Glioma classification: a molecular reappraisal. Am J Pathol 159:779–786
van den Bent MJ, Carpentier AF, Brandes AA et al (2006) Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial. J Clin Oncol 24:2715–2722
Cairncross G, Berkey B et al (2006) Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol 24:2707–2714
Giannini C, Burger PC, Berkey BA et al (2008) Anaplastic oligodendroglial tumors: refining the correlation among histopathology, 1p 19q deletion and clinical outcome in Intergroup Radiation Therapy Oncology Group Trial 9402. Brain Pathol 18:360–369
Giannini C, Scheithauer BW, Weaver AL et al (2001) Oligodendrogliomas: reproducibility and prognostic value of histologic diagnosis and grading. J Neuropathol Exp Neurol 60:248–262
Kros JM, Gorlia T, Kouwenhoven MC et al (2007) Panel review of anaplastic oligodendroglioma from European Organization For Research and Treatment of Cancer Trial 26951: assessment of consensus in diagnosis, influence of 1p/19q loss, and correlations with outcome. J Neuropathol Exp Neurol 66:545–551
Reifenberger J, Reifenberger G, Liu L, James CD, Wechsler W, Collins VP (1994) Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am J Pathol 145:1175–1190
Cairncross JG, Ueki K, Zlatescu MC et al (1998) Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst 90:1473–1479
Ino Y, Betensky RA, Zlatescu MC et al (2001) Molecular subtypes of anaplastic oligodendroglioma: implications for patient management at diagnosis. Clin Cancer Res 7:839–845
Smith JS, Perry A, Borell TJ et al (2000) Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. J Clin Oncol 18:636–645
Jenkins RB, Blair H, Ballman KV et al (2006) A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res 66:9852–9861
Griffin CA, Burger P, Morsberger L et al (2006) Identification of der(1;19)(q10;p10) in five oligodendrogliomas suggests mechanism of concurrent 1p and 19q loss. J Neuropathol Exp Neurol 65:988–994
Idbaih A, Marie Y, Lucchesi C et al (2008) BAC array CGH distinguishes mutually exclusive alterations that define clinicogenetic subtypes of gliomas. Int J Cancer 122:1778–1786
Wen PY, Kesari S (2008) Malignant gliomas in adults. N Engl J Med 359:492–507
Miller CR, Dunham CP, Scheithauer BW, Perry A (2006) Significance of necrosis in grading of oligodendroglial neoplasms: a clinicopathologic and genetic study of newly diagnosed high-grade gliomas. J Clin Oncol 24:5419–5426
Ohgaki H, Dessen P, Jourde B et al (2004) Genetic pathways to glioblastoma: a population-based study. Cancer Res 64:6892–6899
Perry A, Aldape KD, George DH, Burger PC (2004) Small cell astrocytoma: an aggressive variant that is clinicopathologically and genetically distinct from anaplastic oligodendroglioma. Cancer 101:2318–2326
Franco-Hernandez C, Martinez-Glez V, Alonso ME et al (2007) Gene dosage and mutational analyses of EGFR in oligodendrogliomas. Int J Oncol 30:209–215
Nutt CL (2005) Molecular genetics of oligodendrogliomas: a model for improved clinical management in the field of neurooncology. Neurosurg Focus 19:E2
Roerig P, Nessling M, Radlwimmer B et al (2005) Molecular classification of human gliomas using matrix-based comparative genomic hybridization. Int J Cancer 117:95–103
Louis DN, Ohgaki H, Wietsler OD, Cavanee WK (2007) WHO classification of tumors of the central nervous system, 4th edn. International Agency for Cancer Research, Lyon
Fuller CE, Schmidt RE, Roth KA et al (2003) Clinical utility of fluorescence in situ hybridization (FISH) in morphologically ambiguous gliomas with hybrid oligodendroglial/astrocytic features. J Neuropathol Exp Neurol 62:1118–1128
Liva S, Hupé P, Neuvial P et al (2006) CAPweb: a bioinformatics CGH array analysis platform. Nucleic Acids Res 34:W477–W481
Kouwenhoven MC, Kros JM, French PJ et al (2006) 1p/19q loss within oligodendroglioma is predictive for response to first line temozolomide but not to salvage treatment. Eur J Cancer 42:2499–2503
Dalmasso C, Pickrell J, Tuefferd M, Génin E, Bourgain C, Broët P (2007) A mixture model approach to multiple testing for the genetic analysis of gene expression. BMC Proc 1:S141
Zlatescu MC, TehraniYazdi A, Sasaki H et al (2001) Tumor location and growth pattern correlate with genetic signature in oligodendroglial neoplasms. Cancer Res 61:6713–6715
Idbaih A, Marie Y, Pierron G et al (2005) Two types of chromosome 1p losses with opposite significance in gliomas. Ann Neurol 58:483–487
Ichimura K, Vogazianou AP, Liu L et al (2008) 1p36 is a preferential target of chromosome 1 deletions in astrocytic tumours and homozygously deleted in a subset of glioblastomas. Oncogene 27:2097–2108
Jeuken J, Cornelissen S, Boots-Sprenger S, Gijsen S, Wesseling P (2006) Multiplex ligation-dependent probe amplification: a diagnostic tool for simultaneous identification of different genetic markers in glial tumors. J Mol Diagn 8:433–443
Fallon KB, Palmer CA, Roth KA et al (2004) Prognostic value of 1p, 19q, 9p, 10q, and EGFR-FISH analyses in recurrent oligodendrogliomas. J Neuropathol Exp Neurol 63:314–322
Ueki K, Nishikawa R, Nakazato Y et al (2002) Correlation of histology and molecular genetic analysis of 1p, 19q, 10q, TP53, EGFR, CDK4, and CDKN2A in 91 astrocytic and oligodendroglial tumors. Clin Cancer Res 8:196–201
Kouwenhoven MC, Gorlia T, Kros JM et al (2009) Molecular analysis of anaplastic oligodendroglial tumors in a prospective randomized study: a report from EORTC study 26951. Neuro Oncol 11:737–746
Marie Y, Sanson M, Mokhtari K et al (2001) OLIG2 as a specific marker of oligodendroglial tumour cells. Lancet 358:298–300
Ligon KL, Alberta JA, Kho AT et al (2004) The oligodendroglial lineage marker OLIG2 is universally expressed in diffuse gliomas. J Neuropathol Exp Neurol 63:499–509
Cowell JK, Barnett GH, Nowak NJ (2004) Characterization of the 1p/19q chromosomal loss in oligodendrogliomas using comparative genomic hybridization arrays (CGHa). J Neuropathol Exp Neurol 63:151–158
Hui AB, Lo KW, Yin XL, Poon WS, Ng HK (2001) Detection of multiple gene amplifications in glioblastoma multiforme using array-based comparative genomic hybridization. Lab Invest 81:717–723
Rossi MR, Gaile D, Laduca J et al (2005) Identification of consistent novel submegabase deletions in low-grade oligodendrogliomas using array-based comparative genomic hybridization. Genes Chromosomes Cancer 44:85–96
Misra A, Pellarin M, Nigro J et al (2005) Array comparative genomic hybridization identifies genetic subgroups in grade 4 human astrocytoma. Clin Cancer Res 11:2907–2918
Nigro JM, Misra A, Zhang L et al (2005) Integrated array-comparative genomic hybridization and expression array profiles identify clinically relevant molecular subtypes of glioblastoma. Cancer Res 65:1678–1686
Bredel M, Bredel C, Juric D et al (2005) High-resolution genome-wide mapping of genetic alterations in human glial brain tumors. Cancer Res 65:4088–4096
Pfister S, Janzarik WG, Remke M et al (2008) BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest 118:1739–1749
Kotliarov Y, Steed ME, Christopher N et al (2006) High-resolution global genomic survey of 178 gliomas reveals novel regions of copy number alteration and allelic imbalances. Cancer Res 66:9428–9436
Nakahara Y, Shiraishi T, Okamoto H et al (2004) Detrended fluctuation analysis of genome-wide copy number profiles of glioblastomas using array-based comparative genomic hybridization. Neuro Oncol 6:281–289
Kitange G, Misra A, Law M et al (2005) Chromosomal imbalances detected by array comparative genomic hybridization in human oligodendrogliomas and mixed oligoastrocytomas. Genes Chromosomes Cancer 42:68–77
Trost D, Ehrler M, Fimmers R et al (2007) Identification of genomic aberrations associated with shorter overall survival in patients with oligodendroglial tumors. Int J Cancer 120:2368–2376
Idbaih A, Crinière E, Marie Y et al (2008) Gene amplification is a poor prognostic factor in anaplastic oligodendrogliomas. Neuro Oncol 10:540–547
Mohapatra G, Betensky RA, Miller ER et al (2006) Glioma test array for use with formalin-fixed, paraffin-embedded tissue: array comparative genomic hybridization correlates with loss of heterozygosity and fluorescence in situ hybridization. J Mol Diagn 8:268–276
Johnson NA, Hamoudi RA, Ichimura K et al (2006) Application of array CGH on archival formalin-fixed paraffin-embedded tissues including small numbers of microdissected cells. Lab Invest 86:968–978
Maher EA, Brennan C, Wen PY et al (2006) Marked genomic differences characterize primary and secondary glioblastoma subtypes and identify two distinct molecular and clinical secondary glioblastoma entities. Cancer Res 66:11502–11513
Acknowledgments
This work was supported by grants from the INSERM, the Carte d’Identité des Tumeurs CIT) program of the Ligue Nationale Contre le Cancer, the Dutch cancer society.
Conflict of interest
None.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Idbaih, A., Dalmasso, C., Kouwenhoven, M. et al. Genomic aberrations associated with outcome in anaplastic oligodendroglial tumors treated within the EORTC phase III trial 26951. J Neurooncol 103, 221–230 (2011). https://doi.org/10.1007/s11060-010-0380-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-010-0380-9
Keywords
Profiles
- Pim French View author profile