
Abstract
Background
Deafness is the most prevalent human sensorineural defect. It may occur as a result of an external auditory canal involvement, or a deficiency in the sound conduction mechanism, or an impairment of the cochlea, the cochlear nerve or central auditory perception. The genetic causes are the most common, as approximately 70% of hearing disorders are of hereditary origin, divided into two groups, syndromic (associated with other symptoms) and no syndromic (isolated deafness).
Methods
A whole exome sequencing was performed to identify the genetic cause of hearing loss in six Moroccan families and Sanger sequencing was used to validate mutations in these genes.
The results
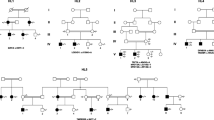
The results of four out of the six families revealed four genetic variants in the genes GJB2, COL4A3, ATP6V1B1 and EDNRB responsible for non-syndromic and syndromic hearing loss. Multiple Bioinformatics programs and molecular modelling predicted the pathogenic effect of these mutations.
Conclusions
We identified in Moroccan deaf patients four homozygous mutations. These results show the importance of whole exome sequencing to identify pathogenic mutations in heterogeneous disorders with multiple genes responsible.


Similar content being viewed by others


Availability of data and material
Data will be provided by the authors upon request.
Code availability
Not applicable.
References
Olusanya BO, Davis AC, Hoffman HJ (2019) Hearing loss: rising prevalence and impact. Bull World Health Organ 97:646–646. https://doi.org/10.2471/BLT.19.224683. A
Wong S-H, Yen Y-C, Li S-Y, Yang J-J (2020) Novel Mutations in the TMPRSS3 Gene may Contribute to Taiwanese Patients with Nonsyndromic Hearing Loss. Int J Mol Sci 21:E2382. https://doi.org/10.3390/ijms21072382
Buonfiglio P, Bruque CD, Luce L et al (2020) GJB2 and GJB6 Genetic Variant Curation in an Argentinean Non-Syndromic Hearing-Impaired Cohort. Genes 11:E1233. https://doi.org/10.3390/genes11101233
Bakhchane A, Bousfiha A, Charoute H et al (2016) Update of the spectrum of GJB2 gene mutations in 152 Moroccan families with autosomal recessive nonsyndromic hearing loss. Eur J Med Genet 59:325–329. https://doi.org/10.1016/j.ejmg.2016.05.002
Charif M, Bounaceur S, Abidi O et al (2012) The c.242G>A mutation in LRTOMT gene is responsible for a high prevalence of deafness in the Moroccan population. Mol Biol Rep 39:11011–11016. https://doi.org/10.1007/s11033-012-2003-3
Bakhchane A, Charif M, Bousfiha A et al (2017) Novel compound heterozygous MYO7A mutations in Moroccan families with autosomal recessive non-syndromic hearing loss. PLoS ONE 12:e0176516. https://doi.org/10.1371/journal.pone.0176516
Bousfiha A, Bakhchane A, Charoute H et al (2017) A novel PEX1 mutation in a Moroccan family with Zellweger spectrum disorders. Hum Genome Var 4:17009. https://doi.org/10.1038/hgv.2017.9
Bousfiha A, Bakhchane A, Charoute H et al (2017) Novel compound heterozygous mutations in the GPR98 (USH2C) gene identified by whole exome sequencing in a Moroccan deaf family. Mol Biol Rep 44:429–434. https://doi.org/10.1007/s11033-017-4129-9
Bakhchane A, Charoute H, Nahili H et al (2015) A novel mutation in the TMC1 gene causes non-syndromic hearing loss in a Moroccan family. Gene 574:28–33. https://doi.org/10.1016/j.gene.2015.07.075
Charif M, Bakhchane A, Abidi O et al (2013) Analysis of CLDN14 gene in deaf Moroccan patients with non-syndromic hearing loss. Gene 523:103–105. https://doi.org/10.1016/j.gene.2013.03.123
Salime S, Charif M, Bousfiha A et al (2017) Homozygous mutations in PJVK and MYO15A genes associated with non-syndromic hearing loss in Moroccan families. Int J Pediatr Otorhinolaryngol 101:25–29. https://doi.org/10.1016/j.ijporl.2017.07.024
Amalou G, Bonnet C, Riahi Z et al (2021) A homozygous MPZL2 deletion is associated with non syndromic hearing loss in a moroccan family. Int J Pediatr Otorhinolaryngol 140:110481. https://doi.org/10.1016/j.ijporl.2020.110481
Shearer AE, Hildebrand MS, Smith RJ (2017) Hereditary Hearing Loss and Deafness Overview. In: Adam MP, Ardinger HH, Pagon RA et al (eds) GeneReviews®. University of Washington, Seattle, Seattle (WA)
Shearer AE, DeLuca AP, Hildebrand MS et al (2010) Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing. Proc Natl Acad Sci U S A 107:21104–21109. https://doi.org/10.1073/pnas.1012989107
Biasini M, Bienert S, Waterhouse A et al (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–258. https://doi.org/10.1093/nar/gku340
Krieger E, Joo K, Lee J et al (2009) Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 77 Suppl 9:114–122. https://doi.org/10.1002/prot.22570
Krieger E, Vriend G (2014) YASARA View - molecular graphics for all devices - from smartphones to workstations. Bioinforma Oxf Engl 30:2981–2982. https://doi.org/10.1093/bioinformatics/btu426
Pires DEV, Ascher DB, Blundell TL (2014) mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinforma Oxf Engl 30:335–342. https://doi.org/10.1093/bioinformatics/btt691
Pandurangan AP, Ochoa-Montaño B, Ascher DB, Blundell TL (2017) SDM: a server for predicting effects of mutations on protein stability. Nucleic Acids Res 45:W229–W235. https://doi.org/10.1093/nar/gkx439
Pires DEV, Ascher DB, Blundell TL (2014) DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res 42:W314–319. https://doi.org/10.1093/nar/gku411
Cao H, Wang J, He L et al (2019) DeepDDG: Predicting the Stability Change of Protein Point Mutations Using Neural Networks. J Chem Inf Model 59:1508–1514. https://doi.org/10.1021/acs.jcim.8b00697
Abidi O, Boulouiz R, Nahili H et al (2008) Carrier frequencies of mutations/polymorphisms in the connexin 26 gene (GJB2) in the Moroccan population. Genet Test 12:569–574. https://doi.org/10.1089/gte.2008.0063
Hamelmann C, Amedofu GK, Albrecht K et al (2001) Pattern of connexin 26 (GJB2) mutations causing sensorineural hearing impairment in Ghana. Hum Mutat 18:84–85. https://doi.org/10.1002/humu.1156
Shi L, Chen J, Li J et al (2016) Prevalence of GJB2 gene mutation in 330 cochlear implant patients in the Jiangsu province. J Laryngol Otol 130:902–906. https://doi.org/10.1017/S0022215116008689
Pavithra A, Chandru J, Jeffrey JM et al (2017) Rare compound heterozygosity involving dominant and recessive mutations of GJB2 gene in an assortative mating hearing impaired Indian family. Eur Arch Oto-Rhino-Laryngol Off J Eur Fed Oto-Rhino-Laryngol Soc EUFOS Affil Ger Soc Oto-Rhino-Laryngol -. Head Neck Surg 274:119–125. https://doi.org/10.1007/s00405-016-4229-5
Boualla L, Jdioui W, Soulami K et al (2016) Clinical and molecular findings in three Moroccan families with distal renal tubular acidosis and deafness: Report of a novel mutation of ATP6V1B1 gene. Curr Res Transl Med 64:5–8. https://doi.org/10.1016/j.retram.2016.01.005
Escobar LI, Simian C, Treard C et al (2016) Mutations in ATP6V1B1 and ATP6V0A4 genes cause recessive distal renal tubular acidosis in Mexican families. Mol Genet Genomic Med 4:303–311. https://doi.org/10.1002/mgg3.205
Elhayek D, Perez de Nanclares G, Chouchane S et al (2013) Molecular diagnosis of distal renal tubular acidosis in Tunisian patients: proposed algorithm for Northern Africa populations for the ATP6V1B1, ATP6V0A4 and SCL4A1 genes. BMC Med Genet 14:119. https://doi.org/10.1186/1471-2350-14-119
Fallerini C, Baldassarri M, Trevisson E et al (2017) Alport syndrome: impact of digenic inheritance in patients management. Clin Genet 92:34–44. https://doi.org/10.1111/cge.12919
Heidet L, Arrondel C, Forestier L et al (2001) Structure of the human type IV collagen gene COL4A3 and mutations in autosomal Alport syndrome. J Am Soc Nephrol JASN 12:97–106
Issa S, Bondurand N, Faubert E et al (2017) EDNRB mutations cause Waardenburg syndrome type II in the heterozygous state. Hum Mutat 38:581–593. https://doi.org/10.1002/humu.23206
Li W, Mei L, Chen H et al (2019) New Genotypes and Phenotypes in Patients with 3 Subtypes of Waardenburg Syndrome Identified by Diagnostic Next-Generation Sequencing. Neural Plast 2019:7143458. https://doi.org/10.1155/2019/7143458
Morimoto N, Mutai H, Namba K et al (2018) Homozygous EDNRB mutation in a patient with Waardenburg syndrome type 1. Auris Nasus Larynx 45:222–226. https://doi.org/10.1016/j.anl.2017.03.022
Acknowledgements
The authors are indebted to the families who contributed to this study. This project was supported by the Institut Pasteur du Maroc (IPM), we also thanks Dr. Snoussi Khalid for the clinical auditory investigation.
Funding
No funds, grants, or other support was received.
Author information
Authors and Affiliations
Contributions
Conceptualization: [Houria Abdelghaffar], [Abdelhamid Barakat] and [Hicham Charoute] ; Formal analysis: [Imane Ait Raise], [Ghita Amalou] and [Amale Bousfiha] ; Funding acquisition: [Abdelhamid Barakat], [Hassan Rouba]; Investigation: [Imane Ait Raise], [Ghita Amalou]; Methodology: [Imane Ait Raise], [Amale Bousfiha] and [Crystel Bonnet] ; Resources: [Christine Petit] and [Abdelhamid Barakat]; Software: [Imane Ait Raise], [Hicham Charoute]; Writing—original draft: [Imane Ait Raise], [Ghita Amalou]; Writing—review & editing: [Houria Abdelghaffar], [Crystel Bonnet], and [Abdelhamid Barakat].
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
The genetic study was approved by the medical ethics committee of the Morocco Pasteur Institute.
Consent to participate
Informed consent was obtained from legal guardians.
Consent to publish
Patients signed informed consent regarding publishing their data.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
AitRaise, I., Amalou, G., Bousfiha, A. et al. Genetic heterogeneity in GJB2, COL4A3, ATP6V1B1 and EDNRB variants detected among hearing impaired families in Morocco. Mol Biol Rep 49, 3949–3954 (2022). https://doi.org/10.1007/s11033-022-07245-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-07245-z