
Abstract
Non-small cell lung cancer (NSCLC) is a widespread and often aggressive form of cancer affecting people worldwide. PIK3CA missense mutations play a significant role in the progression of growth factor signaling in cancer, making PI3Kα an important biological target for inhibition against NSCLC. Natural product molecules with PI3Kα inhibitory activity are promising therapeutic agents for the treatment of NSCLC, owing to their selectivity and potentially lower toxicity compared to synthetic compounds. To discover new natural product molecules, we integrated ligand-based virtual screening with structure-based virtual screening. We developed a multi-ligand pharmacophore hypothesis, validated it with 3D Field-based QSAR, and screened a Natural-Product-Based Library (ChemDiv) containing 3601 molecules. After initial screening, 137 hit molecules were generated and further screened using the extra precision (XP) Glide docking protocol. The best ten molecules were selected for free binding energy (ΔG) analysis using MMGBSA and ADME predictions. For further optimization, the top four hits were subjected to induced fit docking (IFD), quantum chemical descriptors analysis by Frontier Molecular Orbital (FMO) studies, and a 100 ns molecular dynamics (MD) simulation. The compounds—S721-1955, CM4579-5085, S721-1963, and S721-1999—exhibited better results than the PI3Kα selective inhibitor alpelisib. In silico prediction analysis of S721-1955 and alpelisib revealed that the former exhibited superior selectivity theoretically, as evidenced by its higher affinity for the target protein. The selective natural product molecule identified in this study holds promise as a potential anti-cancer drug against NSCLC in the near future, but further in vitro and in vivo studies are necessary to confirm its efficacy.
Graphical Abstract

















Similar content being viewed by others
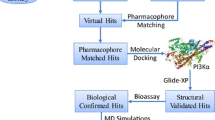
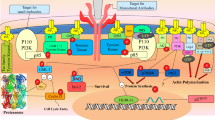
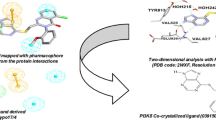
Data availability
The data generated in this study is presented in this manuscript and the additional data is also supplied in the supporting file.
Abbreviations
- Arg:
-
Arginine
- Asn:
-
Asparagine
- Asp:
-
Aspartic acid
- AI:
-
Artificial Intelligence
- DFT:
-
Density Function Theory
- EGFR:
-
Epidermal growth factor receptor
- FMO:
-
Frontier Molecular Orbital
- Glide:
-
Grid-based ligand docking with energetics
- Gln:
-
Glutamine
- Glu:
-
Glutamate
- NSCLC:
-
Non-small cell lung cancer
- His:
-
Histidine
- HTVS:
-
High Throughput Virtual Screening
- IGFR:
-
Insulin-like growth factor receptor
- IL:
-
Interleukin
- INSR:
-
Insulin receptor
- IFD:
-
Induced Fit Docking
- Ile:
-
Isoleucine
- Lys:
-
Lysine
- MD:
-
Molecular Dynamics
- ML:
-
Machine Learning
- Met:
-
Methionine
- mTOR:
-
Mammalian target of rapamycin
- MMGBSA:
-
Molecular Mechanics Generalized Born Surface Area
- OPLS:
-
Optimized potentials for liquid simulations
- PI3K:
-
Phosphatidylinositol 3-kinase
- Phe:
-
Phenylalanine
- Pro:
-
Proline
- QSAR:
-
Quantitative Structure Activity Relationship
- Ras:
-
Rat sarcoma virus
- RMSD:
-
Root mean square deviation
- Ser:
-
Serine
- STAT:
-
Signal transducers and activators of transcription
- SP:
-
Standard precision
- SPC:
-
Simple point charge
- SID:
-
Simulation Interaction Diagram
- SA:
-
Synthetic accessibility
- Thr:
-
Threonine
- Trp:
-
Tryptophan
- Tyr:
-
Tyrosine
- VEGFA:
-
Vascular endothelial growth factor A
- VSGB:
-
Surface Generalized Born Model and Variable Dielectric
- Val:
-
Valine
- XP:
-
Extra precision
References
Paul D, Sanap G, Shenoy S et al (2021) Artificial intelligence in drug discovery and development. Drug Discov Today 26:80–93. https://doi.org/10.1016/j.drudis.2020.10.010
Jana S, Ganeshpurkar A, Singh SK (2018) Multiple 3D-QSAR modeling, e-pharmacophore, molecular docking, and in vitro study to explore novel AChE inhibitors. RSC Adv 8:39477–39495
Cancer Facts & Figures (2023) Atlanta: American Cancer Society, Inc. 2022. In: Am. Cancer Soc. https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/2023-cancer-facts-figures.html
Halder D, Das S, Joseph A, Jeyaprakash RS (2022) Molecular docking and dynamics approach to in silico drug repurposing for inflammatory bowels disease by targeting TNF alpha. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2022.2050948
Halder D, Das S, Aiswarya R, Jeyaprakash RS (2022) Molecular docking and dynamics based approach for the identification of kinase inhibitors targeting PI3Kα against non-small cell lung cancer: a computational study. RSC Adv 12:21452–21467. https://doi.org/10.1039/D2RA03451D
Karakas B, Bachman KE, Park BH (2006) Mutation of the PIK3CA oncogene in human cancers. Br J Cancer 94:455–459. https://doi.org/10.1038/sj.bjc.6602970
Tan AC (2020) Targeting the PI3K/Akt/mTOR pathway in non-small cell lung cancer (NSCLC). Thorac Cancer 11:511–518. https://doi.org/10.1111/1759-7714.13328
Samuels Y, Waldman T (2010) Oncogenic Mutations of PIK3CA in Human Cancers. In: Rommel C, Vanhaesebroeck B, Vogt P. (eds) Phosphoinositide 3-kinase in Health and Disease. Current Topics in Microbiology and Immunology, vol 347. Springer, Berlin, Heidelberg, https://doi.org/10.1007/82_2010_68
Das S, Roy S, Rahaman SB et al (2022) Structure-activity relationship insight of naturally occurring bioactive molecules and their derivatives against non-small cell lung cancer: a comprehensive review. Curr Med Chem. https://doi.org/10.2174/0929867329666220509112423
Yin B, Fang D-M, Zhou X-L, Gao F (2019) Natural products as important tyrosine kinase inhibitors. Eur J Med Chem 182:111664. https://doi.org/10.1016/j.ejmech.2019.111664
Staben ST, Ndubaku C, Blaquiere N et al (2013) Discovery of thiazolobenzoxepin PI3-kinase inhibitors that spare the PI3-kinase β isoform. Bioorganic Med Chem Lett 23:2606–2613. https://doi.org/10.1016/j.bmcl.2013.02.102
(2021) Schrödinger release 2022–4: LigPrep, Schrödinger, LLC, New York
(2023) Schrödinger release 2018–3: Schrödinger, LLC, New York
Lu C, Wu C, Ghoreishi D et al (2021) OPLS4: improving force field accuracy on challenging regimes of chemical space. J Chem Theory Comput 17:4291–4300. https://doi.org/10.1021/acs.jctc.1c00302
Gao Q, Wang Y, Hou J et al (2017) Multiple receptor-ligand based pharmacophore modeling and molecular docking to screen the selective inhibitors of matrix metalloproteinase-9 from natural products. J Comput Aided Mol Des 31:625–641. https://doi.org/10.1007/s10822-017-0028-3
(2021) Schrödinger release 2022–4: field-based QSAR, Schrödinger, LLC, New York
Friesner RA, Banks JL, Murphy RB et al (2004) Glide: a new approach for rapid, accurate docking and scoring: 1—method and assessment of docking accuracy. J Med Chem 47:1739–1749. https://doi.org/10.1021/jm0306430
Halgren TA, Murphy RB, Friesner RA et al (2004) Glide: a new approach for rapid, accurate docking and scoring: 2—enrichment factors in database screening. J Med Chem 47:1750–1759. https://doi.org/10.1021/jm030644s
(2021) Schrödinger release 2023–1: Epik, Schrödinger, LLC, New York
Heffron TP, Heald RA, Ndubaku C et al (2016) The rational design of selective benzoxazepin inhibitors of the α-isoform of phosphoinositide 3-kinase culminating in the identification of ( S )-2-((2-(1-isopropyl-1 H -1,2,4-triazol-5-yl)-5,6-dihydrobenzo [f ]imidazo [1,2- d ] [1,4]oxazepin-9-yl)oxy)propa. J Med Chem 59:985–1002. https://doi.org/10.1021/acs.jmedchem.5b01483
(2021) Schrödinger release 2021–4: protein preparation wizard; Epik, Schrödinger, LLC, New York; impact, Schrödinger, LLC, New York; Prime, Schrödinger, LLC, New York
(2021) Schrödinger release 2023–1: prime, Schrödinger, LLC, New York
Li H, Robertson AD, Jensen JH (2005) Very fast empirical prediction and rationalization of protein pKa values. Proteins Struct Funct Bioinforma 61:704–721. https://doi.org/10.1002/prot.20660
(2021) Schrödinger release 2023–1: glide, Schrödinger, LLC, New York
Li J, Abel R, Zhu K et al (2011) The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins Struct Funct Bioinfo 79:2794–2812. https://doi.org/10.1002/prot.23106
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7:1–13. https://doi.org/10.1038/srep42717
Daina A, Michielin O, Zoete V (2014) ILOGP: A simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J Chem Inf Model 54:3284–3301. https://doi.org/10.1021/CI500467K
(2021) Schrödinger release 2022–4: Jaguar, Schrödinger, LLC, New York
Ye N, Yang Z, Liu Y (2022) Applications of density functional theory in COVID-19 drug modeling. Drug Discov Today 27:1411–1419. https://doi.org/10.1016/j.drudis.2021.12.017
Bouzina A, Berredjem M, Bouacida S et al (2022) Synthesis, in silico study (DFT, ADMET) and crystal structure of novel sulfamoyloxy-oxazolidinones: interaction with SARS-CoV-2. J Mol Struct 1257:132579. https://doi.org/10.1016/j.molstruc.2022.132579
(2021) Schrödinger release 2022–4: QikProp, Schrödinger, LLC, New York
(2021) Schrödinger release 2021–4: desmond molecular dynamics system, D. E. Shaw Research, New York; Maestro-desmond interoperability tools, Schrödinger, New York
Daina A, Michielin O, Zoete V (2019) SwissTargetPrediction: updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res 47:W357–W364. https://doi.org/10.1093/nar/gkz382
Nath V, Ramchandani M, Kumar N et al (2021) Computational identification of potential dipeptidyl peptidase (DPP)-IV inhibitors: structure based virtual screening, molecular dynamics simulation and knowledge based SAR studies. J Mol Struct 1224:129006. https://doi.org/10.1016/j.molstruc.2020.129006
Sándor M, Kiss R, Keserű GM (2010) Virtual fragment docking by glide: a validation study on 190 protein−fragment complexes. J Chem Inf Model 50:1165–1172. https://doi.org/10.1021/ci1000407
Zhu Y-F (2012) PI3K expression and PIK3CA mutations are related to colorectal cancer metastases. World J Gastroenterol 18:3745. https://doi.org/10.3748/wjg.v18.i28.3745
Volinia S, Hiles I, Ormondroyd E et al (1994) Molecular cloning, cDNA sequence, and chromosomal localization of the human phosphatidylinositol 3-kinase p110α (PIK3CA) gene. Genomics 24:472–477. https://doi.org/10.1006/geno.1994.1655
Haider K, Ahmad K, Najmi AK et al (2022) Design, synthesis, biological evaluation, and in silico studies of 2-aminobenzothiazole derivatives as potent PI3Kα inhibitors. Arch Pharm (Weinheim). https://doi.org/10.1002/ardp.202200146
Acknowledgements
The authors would like to express their gratitude to the Manipal-Schrödinger Centre for Molecular Simulations. The authors wish to also thank the Manipal College of Pharmaceutical Sciences for providing the necessary resources for this study. The authors also thank ChemDraw and BioRender.com.
Funding
None.
Author information
Authors and Affiliations
Contributions
D.H. wrote the main manuscript text and prepared figures and tables. S.D. wrote the main manuscript text, prepared figures, and tables and supervised and reviewed the manuscript. J.P. reviewed the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest in this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Halder, D., Das, S. & Jeyaprakash, R.S. Identification of natural product as selective PI3Kα inhibitor against NSCLC: multi-ligand pharmacophore modeling, molecular docking, ADME, DFT, and MD simulations. Mol Divers (2023). https://doi.org/10.1007/s11030-023-10727-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11030-023-10727-2