
Abstract
An immunoinformatics technique was used to predict a monovalent amide immunogen candidate capable of producing therapeutic antibodies as well as a potent immunogen candidate capable of acting as a universal vaccination against all dengue fever virus serotypes. The capsid protein is an attractive goal for anti-DENV due to its position in the dengue existence cycle. The widely accessible immunological data, advances in antigenic peptide prediction using reverse vaccinology, and the introduction of molecular docking in immunoinformatics have directed vaccine manufacturing. The C-proteins of DENV-1-4 serotypes were known as antigens to assist with logical design. Binding epitopes for TC cells, TH cells, and B cells is predicted from structural dengue virus capsid proteins. Each T cell epitope of C-protein integrated with a B cell as a templet was used as a vaccine and produce antibodies in contrast to serotype of the dengue virus. A chimeric tetravalent vaccine was created by combining four vaccines, each representing four dengue serotypes, to serve as a standard vaccine candidate for all four Sero groups. The LKRARNRVS, RGFRKEIGR, KNGAIKVLR, and KAINVLRGF from dengue 1, dengue 2, dengue 3, and dengue 4 epitopes may be essential immunotherapeutic representatives for controlling outbreaks.
Similar content being viewed by others


Introduction
Mosquitoes require a warm, humid environment in order to reproduce, and are hence categorised as tropical and subtropical animals. However, mosquitoes, which number over 3500 species, are distributed everywhere, save in Antarctica (Servadio et al. 2018; Reiter 2001). Chikungunya, dengue, and Zika viruses are mostly transmitted by Aedes aegypti and Aedes albopictus, resulting in numerous virus infections in people. The primary unresolved issue with co-infections is whether infection with two or more viruses might exacerbate illness severity in comparison to single infections (Vogels et al. 2019). In humans, co-infection occurs when a mosquito transmits two or more viruses in the same bite or when two independent mosquitoes transmit different viruses (Magalhaes et al. 2018).
About one billion individuals are infected by vector-borne diseases that lead to over a million deaths per year. Dengue fever (DF) continues to unfold across Asia, the Pacific, America, Africa, and also the Caribbean, touching a complete of 108 countries (CDC 2020). DF, also known as break bone fever, is tropical infectious diseases caused by four serotypes of the dengue virus that are (DENV 1-4) are possibly associated. When the vector comes into contact with an uninfected host, the pathogen is transferred the most common vector-borne disease is that transmitted by mosquitoes, also known as mosquito-borne sicknesses. Immunoinformatics is the use of computing methods and techniques to interpret, generate, and manipulate immunological information (Tong and Ren 2009). The fields of bioinformatics and immunoinformatics are rapidly evolving in terms of the advancement of sophisticated bioinformatics methods that aid researchers in recognizing immunodominant T-cell and B-cell epitopes, making it easier to design contenders for vaccination. The use of an immunoinformatics approach to the logical design of antigens as a B and T cell epitope based popular tetravalent vaccine has steered the current vaccine development. It has a number of advantages over traditional vaccines, including high accuracy in generating a humoral and cellular immune response and high cost efficacy. The epitope based vaccine is also easier to synthesize, purify, store, and treat than traditional vaccines (Oyarzún and Kobe 2016). The genome of the Dengue virus (DEN) contains 10,696 nucleotides. For a single open reading frame, there are 3391 amino acids (Osatomi and Sumiyoshi 1990). RNA viral genome encodes for structural Capsid protein, which aids in core formation by assembling nucleocapsid viral RNA and plays a role in budding and fusion of the virus with the membrane (Kuhn et al. 2002).
The capsid of the Dengue virus is the first viral protein that can be synthesized during translation and found at the end of the genomes 5′ terminus. The C protein is an 11 kDa protein that forms the nucleocapsid by interacting with viral genomic RNA (Sampath and Padmanabhan 2009). The capsid includes a conserved internal hydrophobic fragment that serves as a membrane anchor domain embedded in the endoplasmic reticulum membrane throughout the infection cycle, despite little sequence conservation with other DENV strains. A conserved hydrophobic domain of Protein C links it to intracellular membranes. Dengue virus-infected cells cytoplasm was used as a factory for capsid protein at the edges of lipid droplets, which are endoplasmic reticulum-derived organelles (Carvalho et al. 2012; Samsa et al. 2009). The number of lipoid droplets per cell inflated throughout infectious disease virus infection, implying a connection between viral replication and lipid-droplet metabolism (Table 1).
Because of its importance within the DF life cycle, the capsid macromolecule is a tempting choice for anti-DENV small molecules. This reasons steric quandary and systemic rigidity, stopping infectious virions from entering, collecting, or releasing. Here we'll look at how capsid can be taken into consideration into vaccine production, especially for monovalent and chimeric tetravalent dengue vaccines many mutations within the capsid macromolecule sequence end in the event of sub infective agent particles. These sub infectious agent particles are immunogenic as a result of they embody the viral surface proteins E and M, however they're not infectious because the viral ordering isn't assembled. As a result, they could be used as vaccines. Tick-borne encephalitis virus was the first flavivirus to show this (Fig. 1).

Population coverage of epitope LKRARNRVS
The adaptive immune system, humoral response performs a significant shielding position in DENV infection. While T cells play an important role in fighting viral infections, they have been linked to both pathological and protective effects in the sense of DENV infection (John and Rathore 2019). DENV CD8+ and CD4+ cells have been shown to play a significant role in DENV infection control in studies. The most important external structures of B-cells responsible for this are cells that promote memory reactions, cell activation, antigen recognition, and signal transduction (Duan et al. 2015; Wahala 2011; Sathe and Cusick 2020). B cells antigen receptor is a functional component of multi molecular protein complexes on the cell surface. CD4 T cells and CD8 T cells have been shown in animal studies to protect against DENV infection (Zellweger et al. 2014). When a T cell recognizes antigen on the surface of a B cell, the T cell becomes stimulated, and then stimulates the B cell. If a B cell is activated, it undergoes clonally expansion, and daughter cells divide into plasma cells. T cell support consists of two components: lymphokines, which function as growth and differentiation factors for B cells. Plasma cells produce massive amounts of antibodies and function as antibody factories (Fig. 2).

Population coverage of epitope RGFRKEIGR
Peptides gift the most important organic phenomenon advanced (MHC) molecules, additionally called human white blood cell substance molecules in humans that used for identification by then lymph cell receptor as a fusion protein. Furthermore, HLA alleles linked to defense against extreme dengue disease are also linked to robust and multifunctional T cell responses, implying that T cells play a defensive role during DENV infection (Grifoni et al. 2017). T cell epitopes can play a role in a serotype-specific or cross-reactive reaction. The B-cell antigen receptor is a transmembrane receptor that crosses into the cytoplasm. These are inefficient at communicating signals and activating B cells (Tanaka and Baba 2020). The BCR (B cell receptor) is a multi-molecular protein complex that is not covalently bound to other proteins. So BCR is needed functionally (Brezski and Monroe 2008).
Dengvaxia® is a chimeric vaccine that protects against both yellow fever and DF. It had a poor overall effectiveness against DENV, with nearly 50% effectiveness against DENV 1 and 39% effectiveness against DENV 2. Latest clinical trials, however, have discovered that the CYDTDV vaccine causes a high risk of hospitalization in children under the age of nine (Hadinegoro et al. 2015). There's a chance that a better result would help for DHF/DSS (Rothman 2004). The main advantages of this epitope-based tetravalent vaccine are the reduced disease incidence and the lack of pathogen interference in the absence of any live parts, since these are specific peptide sequences that can be developed in the in the lab. Protein synthesis and purification methods could be used to obtain the pure form of proposed vaccine structures (Fig. 3).

Population coverage of epitope KNGAIKVLR
DENV infections result in the production of neutralising antibodies (NAbs), which are associated with protection. Numerous DENV vaccine candidates are in various phases of clinical development (de Alwis et al. 2011; Guzman et al. 2007; Mathew et al. 2011). While the existence of NAbs as a correlate of protection has led the development of DENV vaccines, new research reveals that the presence of NAbs to the four serotypes following vaccination is not a reliable predictor of protection (Biswal et al. 2020; Dayan et al. 2020; Moodie et al. 2018). Dengvaxia is a live attenuated chimeric tetravalent dengue vaccine (CYD-TDV) produced by altering the live attenuated yellow fever 17D vaccine to include the envelope (E) and premembrane proteins of each DENV serotype (Thomas and Yoon 2019).
Methodology
Viral Protein Selection for Preparation of Vaccine
To predict the most successful DENV applicants for vaccination, this analysis used statistical methods. The Dengue virus amino acid sequence was obtained from the virus pathogen resource sequence database (https://www.viprbrc.org/brc/vipr-protein-serch.spg). FASTA format was used to extract viral proteins (Fig. 4).

Population coverage of epitope KAINVLRGF
Allergenicity of Protein Predictions
The Allergen FP algorithm, which was stated in the current research, was added (FP stands for Finger Print). Allergen FP is written in Python. It can be found online at http://ddg-pharmfac.net/Allergen FP. Proteins that aren't allergens were chosen based on their similarity index. As contrasted to known allergens, a protein is considered a possible allergen if it has > 35% sequence similarity over an 80-amino-acid window (Stadler and Stadler 2003).
Prediction of Epitopes from Shortlisted Proteins
The IEDB tool is used to identify the most promiscuous epitopes binding to the MHC class I allele. T-cell epitopes, which play a vital function in vaccine design, set off the immune response. The neural network's ability to be trained on data consisting of continuous binding affinities, improves the efficiency of the new system. T cells do this by detecting peptides bound to MHC receptors (major histocompatibility complex). Prediction methods based on alignments of insertions and deletions work considerably better than methods trained on single-length peptides.The position of deletions will help explain the peptide–MHC binding modes, As previously defined, the approach was implemented as a feed-forward artificial neural network ensemble with a single hidden layer (Nielsen and Lund 2009) (IEDB-AR, http://tools.iedb.org).
Predictions of MHC class II binding are commonly used to find epitope candidates in infectious agents. To date, the vast majority of human MHC class II prediction algorithms have focused on HLA molecules encoded in the DR locus. Immune epitopes are HLA class II peptide ligands that are recognised by T cells and trigger an immune response (Mith-Garvin et al. 2009). The immune Epitope Database's unique peptide binding specificity can be influenced by both alpha and beta chains (IEDB) (Vita et al. 2010; Peters and Sette 2007).
Instability and GRAVY of Epitope Prediction
Prot Param is software that measures physical and chemical parameters such as instability for a protein contained in Swiss-Prot or TrEMBLL, as well as for a user-entered epitope sequence (Boeckmann et al. 2003) that is a curate protein sequence database that aims to include high-quality annotations (http://www.expasy.org/sprot/).
The grand average of hydropathicity (GRAVY) comparison of the most abundant epitope in the extracellular matrix of corneal stroma between different species with the Grand Average Hydropathicity (GRAVY) values is one of the computed parameters. We've used this method to choose studies with a higher negative score (Table 2).
Toxicity, Hydrophobicity and Hydropathicity Prediction
Toxin Pred was used to assess epitope toxicity, hydrophobicity, and hydropathicity (Gupta et al. 2013). Toxin Pred is a kind in silico method for predicting peptide toxicity, hydrophobicity, and hydropathicity. It can be used to create the least toxic peptides and find toxic regions of proteins. Toxin Pred (http://crdd.osdd.net/raghava/toxinpred/) can improve peptide-based drug discovery (Table 3).
B-Cell Epitope Prediction
For all stable structural capsid proteins, the ABCpred Prediction Server was used to forecast B-cell epitope linear B-cell epitopes. The predicted epitopes were then shortlisted based on their prediction score. The aim of the ABCpred server is to use an artificial neural network to predict B cell epitope in an antigen sequence.This is the first server to use fixed length patterns and a recurrent neural network (Saha and Raghava 2006).
Consensus Epitope, Immunogenicity and Antigenic Propensity Prediction
The prediction of common epitopes between or within known serotypes may be used to produce Dengue virus monovalent and chimeric tetravalent vaccines. When the findings of the predicted dengue virus 1-4 serotype epitopes were compared, it was discovered that the standard peptides were consensus epitopes. The main reason for using the consensus epitope technique was to sort out potential candidates that were most likely to elicit a Dengue virus immune response (Table 4).
To be stimulated and evoke their effectors roles, T-cells must identify peptides posed on MHC molecules. Several researches suggest that some peptides are more immunogenic than others, indicating that they are more likely to be T-cell epitopes. The discovery of factors that affect immunogenicity would be a crucial next step in the study of T-cell epitopes and our knowledge of cellular immune responses (Calis et al. 2013). To be stimulated and evoke their effectors functions, T cells must recognize peptides expressed on MHC molecules in order to be activated and elicit their effectors functions (Table 5).
Kolaskar and Tongaonkar's method is used to determine antigenic peptides. The predictions are based on a table that shows the frequency of amino acid residues in segmental epitopes that have been studied experimentally (Table 6).
IC50 and Conservancy Analysis
Prediction server Propred (Singh and Raghava 2003) predicted the corresponding allele for each of the proposed T-cell epitopes based on IC50 values. In the in silico vaccinology methodology, conservancy analysis is used to determine the degree of epitope distribution in a homologous protein set. The epitope conservancy research method is for determining the conservation of epitopes (http://tools.iedb.org/conservancy/) at the IEDB was used to forecast the conservancy trend of the target epitopes. We used a method to help with the optimal degree of conservation in epitope selection and to assess the variability of epitopes within a series of protein sequences. Conservancy can be measured using given identity parameters, and minimum and maximal conservancy amounts can be determined.
Population Coverage Analysis
For successful vaccination, a vaccine molecule must have broad-spectrum defense against the disease in various world populations (Robinson et al. 2016). However, due to the strong polymorphism of MHC molecules, individuals of different ethnicities/countries have different MHC related pools/frequencies. This issue was resolved for analysis using the IEDB population coverage method (http:/tools.iedb.org/population/).
The fraction of people who are most likely to react to a given epitope range, vaccine, was calculated using an algorithm based on HLA genotypic frequencies. We used a web-based method to forecast population coverage of T-cell epitope-based diagnostics and vaccines. As a result, epitope-based vaccinations or diagnostics may be optimized to optimize population distribution while minimizing uncertainty and heterogeneity in coverage achieved or predicted across ethnic groups.
Modeling, Refining, and Validation of Tertiary Structures
The PEPstr server predicts the tertiary structure of small peptides with sequence lengths ranging from Residues range from 7 to 25. This is a server that models small peptide structures in three dimensions (http://www.imtech.res.in/raghava/pepstr/).
Structure-Based Modeling and Evaluation of MHC Alleles
Allele structures are prepared using the IMGT/HLA Database (http://www.ebi.ac.uk/ipd/imgt/hla/intro.html). The Anthony Nolan Research Institute's HLA Informatics department has launched the IMGT/HLA Database. The member libraries of the International Nucleotide Sequence Database Collaboration provide access to all of the submissions submitted by the IPD-IMGT/HLA database (Kodama et al. 2019; Sayers et al. 2019).
Computational approaches were used to model some of the allele configurations that were not present in the IMGT/HLA database. Homology simulation modeling technique was used to construct the allele structures. Procheck (http://www.biochem.ucl.ac.uk/) is software that evaluates the stereochemical content of a protein structure by comparing it to well-refined structures of the same resolution and indicating its local, residue-by-residue consistency.
Docking
Signal transduction, immune responses, and cellular modulation are all examples of peptide–protein interactions play an important role (Petsalaki and Russell 2008; London et al. 2013). The composition of protein–peptide complexes must be determined in order to understand the molecular dynamics of related biological processes and to manufacture peptide vaccines. Protein–peptide docking predicts the complex structure from the arrangement of proteins and a sequence of peptides by sampling possible peptide binding conformations and ranking the putative protein–peptide complexes with an energy scoring feature (htt:huanglab.phys.hust.edu.cn/hpepdock/).
HPEPDOCK is a web based server that uses a hierarchical algorithm to perform blind protein–peptide docking. HPEPDOCK considers peptide versatility by an ensemble of peptide conformations created by our MODPEP software, rather than running lengthy simulations to refine peptide conformations (Fosgerau and Hoffmann 2015).
Results and Discussion
Selection of Viral Proteins for Vaccine Preparation
The virus pathogen resource sequence database was used to produce the tetravalent vaccine (https://www.viprbrc.org/brc/vipr-protein-serch.spg). Dengue virus 1-4 was used to determine the amino acid sequence of the structural Capsid protein. The structural protein assists the virus in invading the host and assembling viral particles. The C-proteins of DENV 1-4 Flavivirus serotypes had been selected for this analysis and an epitope was designed in such a way that it generated both B and T cell immunity.
Protein Predictions for Allergenicity
The algorithm described in this study was released on a specially designed website known as Allergen F P. Following the allergenicity test, four non-allergenic capsid proteins were selected for further research based on the index of similarity.
Epitope Estimation for Cytotoxic T Lymphocytes (CTL)
The most promiscuous epitopes absolute to the MHC category I allelomorph for capsid super molecule were classified victimization CTL epitopes projected from the IEDB tool. The immune response is triggered by T cell epitopes. The IEDB server assigns a score to each epitope. A high ranking indicates high precision. A high score indicates that the binder is of high quality.
All epitopes non allergenic structural capsid proteins together with longer chain of peptides, the higher the number of epitopes, based on a higher score of more than 0.1. As a result, for dengue 1, dengue 2, dengue 3, and dengue 4, a total of 29, 69, 40, and 53 CD8+ T cell epitopes were selected in my study.
Helper T Lymphocyte (HTL) Epitope Prediction
For structural capsid protein, the IEDB server for MHC II predicted HTL epitopes. IEDB recommended 2.22 as a prediction process, with Low adjusted rank binders being strong binders. Actual Score 30 is the cutoff value. The epitopes with a score of more than 30 were short out from my data. For dengue 1, dengue 2, dengue 3, and dengue 4, a total of 34, 55, 29, and 47 CD4+ T cell epitopes were eliminated based on a lower score of less than 30 for next study.
Instability and GRAVY of the Epitope Predictions
Prot Param is a way of calculating an index of parameter instability. The epitope is stable if the value is less than 40. Calculate the GRAVY value for hydropathy in protein sequences. The GRAVY value is calculated by multiplying the hydropathy values of all amino acids by the epitope length. Analysis chooses a higher negative score for the next study. On the basis of stability index and negative GRAVY value, 5 epitopes for dengue 1 were shorted out, 20 epitopes for dengue 2, 4 epitopes for dengue 3, and 10 epitopes for dengue 4 were shorted out as CTL epitopes. On the other hand, HCL epitopes are stable and had a negative GRAVY value. 1 epitope, 13 epitopes, 1 epitope and 7 epitopes short out from Dengue 1, Dengue 2, Dengue 3 and Dengue 4.
Toxicity, Hydrophobicity, Hydropathicity prediction
The toxicity of epitopes was determined using the Toxin Pred program, which classified them as toxic or non toxic. Only the negative SVM score indicates that chosen epitopes were non toxic, hydrophobic, and hydropathic, indicating that they should be investigated further study.
Prediction of B-Cell Epitope Vaccine Sequence Construction
All epitopes expected via way of means of ABCpred with a score more than 0.50 have been selected for every capsid protein. From all capsid proteins, a total of 25, 25, 25, and 20 B cell epitopes are predicted for dengue 1, dengue 2, dengue 3, and dengue 4, respectively. Predicted B-cell epitopes were used as a basis for CTL and HTL epitopes in the development of the final vaccine, and those epitopes whose sequences in B cell epitopes overlapped were shortlisted and chosen for inclusion in the vaccine's final build.
Prediction of Consensus Epitope, Immunogenicity, and Antigenic Propensity
A total of 47 consensus epitopes were predicted in my study. For break bone fever dengue 1, dengue 2, dengue 3, and dengue 4, consensus epitopes of 8, 18, 8, and 13 are expected. The immunogenicity and average antigenic propensity of these epitopes were considered for further investigation. Highly antigenic Consensus B cell epitopes were chosen from among B cell epitopes to predict T cell epitopes.
We constructed peptide datasets with equivalent MHC binding affinity and distinguished those that were recognized by T cells from those that were not. T cells with a high Immunogenicity score are more likely to be identified, whereas those with a negative score are less likely to be accepted. For dengue 1, LKRARNRVS had a higher score of 0.07259, RGFRKEIGR and VLRGFRKEI had a higher score of 0.08016 and 0.23167, respectively, for dengue 2, and KNGAIKVLR had a higher score of 0.00533 for dengue 3. For dengue 4, the epitopes KAINVLRGF and INVLRGFRK have higher scores of 0.12993 and 0.21358, respectively. Epitopes that failed to generate a positive value were discarded for further investigation.
The vaccine construct's antigenicity probability was determined to be 0.4 by ANTIGENpro, indicating that it is antigenic. LKRARNRVS has a higher dengue 1 score of 0.9834, RGFRKEIGR and VLRGFRKEI have a higher dengue 2 score of 0.9515 and 1.0022, respectively, and KNGAIKVLR has a higher dengue 3 score of 1.0258. For dengue 4, KAINVLRGF and INVLRGFRK have better scores of 1.178 and 1.0186 respectively. Both results suggest that the final vaccine construct is a powerful antigen as a result of this process.
IC50 Values and Conservancy Prediction Through Consensus Sequences
Peptides with IC50 values of 50 nM are thought to have a high affinity, 500 nM to be intermediate and 5000 nM to be weak in the Informatics in Medicine Unlocked 20 (2020) 1004306 IEDB. Alleles with IC50 values were chosen as the best binders for further investigation.
Three out of six epitopes demonstrated 100% conservation in all consensus sequences of all DENV at the 60% sequence identity threshold. In my research, we discovered that the epitope LKRARNRVS of DENV-1 is 60% conserved, while the epitopes RGFRKEIGR and VLRGFRKEI of DENV-2 are 98.94% and 100.00% conserved, DENV-3 epitope KNGAIKVLR is 97.00% conserved, while DENV-4 epitopes KAINVLRGF and INVLRGFRK are also 100.00% conserved respectively.
Population Coverage Study
Selecting a group of epitopes with many HLA binding capacities will help to expand global coverage. Exploitation HLA constitution frequencies; we have a tendency to be ready to establish the response of every human fraction to a given epitope. In my research, we discovered that the DENV-1 epitope LKRARNRVS covers 76.04% of the population in India, while the average population coverage is 66.49%, and the DENV-2 epitopes RGFRKEIGR and VLRGFRKEI cover 60.69% and 39.65% of the population in India, respectively, while the average population coverage is 98.64% and 47.95%. DENV-3 epitope KNGAIKVLR covered 78.64% of the population in India, while the average population coverage was 68.45%, and DENV-4 epitopes KAINVLRGF and INVLRGFRK covered 78.62% and 76.04% of the population in India, respectively, while the average population coverage was 70.02% and 66.49%. The DENV-1 epitope LKRARNRVS, DENV-2 epitope RGFRKEIGR, DENV-3 epitope KNGAIKVLR, and DENV-4 epitope KAINVLRGF were chosen for further study due to their higher population cover.
Tertiary Structure Modeling, Refinement, and Evaluation
The method makes use of both PSIPRED's projected standard secondary structure information and Beta Turns' predicted information. A typical backbone-dependent rotamer library is used to position the side-chain angles.
Allele Structure
The IPDIMGT/HL allele Structure Prediction server was used to build the 3D structure of the chosen allele HLA B*2705 (2BSR).Using the IPD IMGT/HLA allele Structure Prediction server, the 3D structure of the selected allele DRB1 1501 (1BX2) was created. MODELLER 9.10 is homology simulation software chosen Sample Template (PDB ID) 3PDO by the model with allele DRB1 0701. The model with allelomorph DRB1 1327 selected model (PDB ID) 2WBJ by MODELLER 9.10. PROCHECK was used to check the allele's stereo chemical properties.
Docking
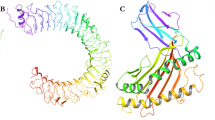
The Hpepdock online server, which was chosen for protein docking, was used for blind global peptide docking to measure the relationship between the refined model and the immune receptor. The success of Hpepdock docking generates a large number of outcomes, from which the top ten had been selected for analysis. After analyzing all 10 docked conformations, result number one was the best-docked model, indicating the best interactions between the receptor and ligand (Fig. 5). Peptide LKRARNRVS, RGFRKEIGR, KNGAIKVLR and KAINVLRGF for DENGUE-1, DENGUE-2, DENGUE-3 and DENGUE-4 bind with Allele DRB1_1327, HLA-B*2705, DRB1_0701 and DRB1_1501 Docking score − 213.922, − 222.148, − 206.13, − 178.031 were found to be geometric form complexity docking score.

Graphical representation of the D1 T-cell epitope (LKRARNRVS) docked with DRB1_1327, D2 T-cell epitope (RGFRKEIGR) docked with HLA-B*2705, D3 T-cell epitope (KNGAIKVLR) docked with DRB1_0701 and D4 T-cell epitope (KAINVLRGF) docked with DRB1_1501
Discussion
DF has emerged as a life-threatening disease that has spread exponentially across the world in recent years. Many efforts have been made to develop the dengue virus vaccine, but none have been effective. Dengvaxia was the first vaccine approved, but it has a number of flaws that make it useless against all DEN serotypes. According to the WHO, Dengvaxia can only be used in patients aged 9 to 45 years old and in countries where DF is widespread. Dengvaxia's ineffectiveness and shortcomings necessitate the creation of a more effective dengue vaccine right away. Using an immunoinformatics approach, an epitope-based tetravalent DENV vaccine was created to provide increased protection over previously manufactured vaccines.
To anticipate capsid protein epitopes, the entire DENV viral proteome was retrieved from the VPRS database. Capsid's role is to bundle the viral genome so that it can be efficiently transferred from one host cell to another. Capsid will form the nucleocapsid before the processing of assembly or packaging faulty viruses, which has become a significant focus of vaccine development strategy (Patkar et al. 2007). It is contained in lipid droplets that form as a result of interactions with host proteins and other factors (Martins et al. 2012; Byk and Gamarnik 2016; Iglesias et al. 2015; Samsa et al. 2009). The dengue C protein is important in virus assembly to ensure precise encapsidation of the viral genome. The presence of sub viral particles emitted from infected cells shows Capsid's essential function currently, information from the capsid structures is used to explain the role of flavivirus capsid in the virus life cycle (Alvarez et al. 2005; Doklandet al. 2004). Encapsidation’s function is unclear, although it is believed to include non structural viral proteins as well as C protein (Villar et al. 2015). In order to find possible drug targets (Oliveira, et al. 2017; Byrd et al. 2013; Soto-Acosta et al. 2014). As the viral polyprotein is cleaved by signal peptidase, the capsid may interact with vesicular transport proteins and pass into lipid droplets for storage or the nucleus (Hasan et al. 2018). Now that established, capsid proteins play a variety of other roles and interact in the pathogenesis of these viruses (Slomnicki et al. 2017; Faizan et al. 2016). Capsid contact with lipid droplet membranes aids in both viral packing and storage capsid prior to packaging (Martins et al. 2012; Samsa et al. 2009; Shang et al. 2018). Capsid of flaviviruses appears to bind nucleic acids (Byk et al. 2016; Samuel et al. 2016; Varjak et al. 2017) in a non-specific manner. Capsid isn't the only soluble viral protein that can bind double-stranded nucleic acids (Cortese et al. 2017; Paul and Bartenschlager 2015). Despite the fact that different types of host proteins have been shown to interact with the capsid protein and despite the fact that certain medications have been engineered to target these interactions. However, it's possible that during the compacting of the genome, the capsid binds both ss- and dsRNA. As a result, a comparison of the nucleic acids that the capsid binds in vivo is needed (Garcia-Blanco et al. 2016).
Antibodies, B cells, and T cells accept the epitope as a component of an antigen that is recognised by the immune system. MHC binding estimates are now highly precise, covering the majority of established HLA specificities. These structures are excellent for epitope exploration (Hope and McLauchlan 2000).
Experimentally determining B cell epitopes is costly in terms of resources and time (Thiele and Spandl 2008). In my research, the ABCpred method was used to estimate B cell epitopes. B cell epitopes, which can only cause B cell mediated immunity, are expected at first. Highly antigenic Consensus B cell epitopes were chosen from among B cell epitopes to predict T cell epitopes.
Many researchers have recently looked at B cell epitope sequences in order to predict T cell epitopes that could interfere with both MHC groups with the most alleles, resulting in a stronger antigenic response (Boulant et al. 2007). Docking the receptor ligand molecule is an effective technique for determining the receptor's relative binding affinity for the ligand. Molecular docking of the expected immunogenic epitope LKRARNRVS (DRB1 1327) of DENV-1, epitope RGFRKEIGR (HLAB*2705) of DENV-2, epitope KNGAIKVLR (DRB1 0701) of DENV-3, and epitope KAINVLRGF (DRB1 1501) of DENV-4 Docking score − 213.922, − 222.148, − 206.13, − 178.031 was performed to suggest structural insight into the epitope. The constructs were found to be effective after testing antigenicity, solubility, and allergenicity. These T cell epitopes may be used as a candidate for a monovalent vaccine that would be safe and immunogenic against DENV serotypes.
A tetravalent dengue DNA vaccine was created by combining four monovalent vaccines in a non-lipid adjuvant and testing it in rhesus monkeys. Antibodies of all four dengue serotypes may be neutralized by the vaccine (Zhu et al. 2007). A dengue vaccine would elicit defensive immune responses to all four serotypes of the disease. In the current study, a monovalent 9-mer epitope induces immunity against a single DENV serotype. This tetravalent vaccine should protect against all four dengue serotypes.
Similar studies were performed by different researchers such as Ali et al. (2017) employed immunoinformatics to develop a multi-epitope-based dengue subunit vaccine capable of eliciting a variety of immune responses within the host. For dengue virus structural and non-structural proteins, distinct B-cell, TC cell, and TH cell binding epitopes were predicted. Final vaccine constructs include epitopes from TC and TH cells and an adjuvant (β-defensin) at the N-terminus. Subramaniyan et al. (2017) investigated E-proteins from DENV-1-4 serotypes as antigens and hypothesised that T cell and B cell epitopes could act as peptide vaccine candidates. The chimeric tetravalent vaccine was developed by conjugating four vaccines, one from each of the four dengue serotypes, to produce a vaccine candidate that is effective against all four dengue serotypes. They determined that the monovalent 9-mer T cell epitopes for each DENV serotype can be exploited to generate unique antibodies against dengue virus, as well as a chimeric common tetravalent vaccine candidate capable of covering any of the four dengue virus serotypes. Verma et al. (2019) conducted a comparative genomes study of Dengue virus (DENV) to identify novel vaccine targets. They discovered 100% conserved epitopes in the envelope protein (RCPTQGE), the NS3 (SAAQRRGR, PGTSGSPI), the NS4A (QRTPQDNQL), the NS4B (LQAKATREAQKRA), and the NS5 (QRGSGQV) proteins in all DENV serotypes. Additionally, conserved serotype-specific motifs were discovered in NS1, NS5, Capsid, PrM, and Envelope proteins. Chan et al. (2020) developed four multi-epitope peptides (P1–P4) against all four DENV serotypes by connecting a universal T-helper epitope (PADRE or TpD) to a highly conserved CD8 T cell epitope and a B cell epitope (B1 or B2). Four nanovaccines (NP1–NP4) were developed using the multi-epitope peptides conjugated to polystyrene nanoparticles (PSNPs). They concluded that NP3, which contained the TpD T-helper epitope coupled to the highly conserved B1 epitope derived from the E protein, was capable of eliciting large quantities of IFN-γ and NAbs against all four dengue serotypes.
Similar studies were performed on different viruses by different researchers such as Pandey et al. (2018) developed a multiepitope subunit vaccine employing Zika virus structural and nonstructural proteins using a combinatorial immunoinformatics method. The subunit vaccine is composed of cytotoxic T-lymphocyte and helper T-lymphocyte epitopes, as well as an adjuvant and linkers. Shahid et al. (2020) developed a MEBP vaccine using a combination of immunoinformatics and molecular docking. Prediction of B-cell, T-cell and IFN-epitopes was performed using the ZIKV proteome. Tahir ul Qamar et al. (2018) conducted a study to identify conserved B and T cell epitopes on CHIKV structural proteins using immunoinformatics and computational techniques. These epitopes may play a critical role in eliciting immunological responses against CHIKV. Numerous conserved CTL epitopes, linear and conformational B cell epitopes, and their antigenicity were predicted for the CHIKV structural polyprotein.
Conclusion
DENV-1 epitope LKRARNRVS, DENV-2 epitope RGFRKEIGR, DENV-3 epitope KNGAIKVLR, and DENV-4 epitope KAINVLRGF were selected as the top four T cell epitopes. Chosen as monovalent vaccines were conjugated to create a compound tetravalent vaccine in this research. Further wet-lab replication using cell-based approaches and animal-based models will boost the credibility of this study.
References
Ali M, Pandey RK, Khatoon N et al (2017) Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci Rep 7:9232. https://doi.org/10.1038/s41598-017-09199-w
Alvarez DE, De Lella EAL, Fucito S, Gamarnik AV (2005) Role of RNA structures present at the 3′-UTR of dengue virus on translation, RNA synthesis, and viral replication. Virology 339:200–212. https://doi.org/10.1016/j.virol.2005.06.009
de Alwis R et al (2011) In-depth analysis of the antibody response of individuals exposed to primary dengue virus infection. PLoS Negl Trop Dis 5(6):e1188
Biswal S et al (2020) Efficacy of a tetravalent dengue vaccine in healthy children aged 4–16 years: a randomised, placebo-controlled, phase 3 trial. Lancet 395(10234):1423–1433
Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O’Donovan C, Phan I et al (2003) The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 31:354–370. https://doi.org/10.1093/nar/gkg095
Boulant S, Targett-Adams P, McLauchlan J (2007) Disrupting the association of hepatitis C virus core protein with lipid droplets correlates with a loss in production of infectious virus. J Gen Virol 88:2204–2213. https://doi.org/10.1099/vir.0.82898-0
Brezski RJ, Monroe JG (2008) B-cell receptor. Adv Exp Med Biol 640:12–21. https://doi.org/10.1007/978-981-15-3532-1_2
Byk LA, Gamarnik AV (2016) Properties and functions of the dengue virus capsid protein. Annu Rev Virol 3:263–281. https://doi.org/10.1146/annurev-virology-110615-042334
Byrd CM, Dai D, Grosenbach DW, Berhanu A, Jones KF, Cardwell KB, Schneider C, Wineinger KA, Page JM, Harver C et al (2013) a novel inhibitor of dengue virus replication that targets the capsid protein. Antimicrob Agents Chemother 57:15–25. https://doi.org/10.1128/aac.01429-12
Calis JJA, Maybeno M, Greenbaum JA, Weiskopf D, De Silva AD, Sette A, Kesmir C, Peters B (2013) Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput Biol 8(1):361. https://doi.org/10.1371/journal.pcbi.1003266
Carvalho FA et al (2012) Dengue virus capsid protein binding to hepatic lipid droplets (LD) is potassium ion dependent and is mediated by LD surface proteins. J Virol 86:2096–2108. https://doi.org/10.1128/JVI.06796-11
Centers for Disease Control and Prevention, CDC (2020). https://www.cdc.gov/dengue/statistics-maps/2020.html
Chan Y, Jazayeri SD, Ramanathan B, Poh CL (2020) Enhancement of tetravalent immune responses to highly conserved epitopes of a dengue peptide vaccine conjugated to polystyrene nanoparticles. Vaccines (basel) 8(3):417. https://doi.org/10.3390/vaccines8030417
Cortese M, Goellner S, Acosta EG, Neufeldt CJ, Oleksiuk O, Lampe M, Haselmann U, Funaya C, Schieber N, Ronchi P et al (2017) Ultrastructural characterization of Zika virus replication factories. Cell Rep 18:2113–2123. https://doi.org/10.1016/j.celrep.2017.02.014
Dayan GH et al (2020) Efficacy after 1 and 2 doses of CYD-TDV in dengue endemic areas by dengue serostatus. Vaccine 38(41):3472–6477
Dokland T, Walsh M, Mackenzie JM, Khromykh AA, Ee KH, Wang S (2004) West Nile virus core protein; tetramer structure and ribbon formation. Structure 12:1157–1163. https://doi.org/10.1016/j.str.2004.04.024
Duan ZL, Liu HF, Huang X, Wang SN, Yang JL, Chen XY et al (2015) Identification of conserved and HLA-A*2402-restricted epitopes in dengue virus serotype2. Virus Res 196:5–12. https://doi.org/10.1016/j.virusres.2014.10.022
Faizan MI, Abdullah M, Ali S, Naqvi IH, Ahmed A, Parveen S (2016) Zika virus-induced microcephaly and its possible molecular mechanism. Intervirology 59:152–158. https://doi.org/10.1159/000452950
Fosgerau K, Hoffmann T (2015) Peptide therapeutics: current status and future directions. Drug Discov Today 20:122–128. https://doi.org/10.1016/j.drudis.2014.10.003
Garcia-Blanco MA, Vasudevan SG, Bradrick SS, Nicchitta C (2016) Flavivirus RNA transactions from viral entry to genome replication. Antivir Res 134:244–249. https://doi.org/10.1016/j.antiviral.2016.09.010
Grifoni A, Angelo MA, Lopez B, O’Rourke PH, Sidney J, Cerpas C et al (2017) Global assessment of dengue virus-specific CD4(+) T cell responses in dengue-endemic areas. Front Immunol 8:1309. https://doi.org/10.3389/fimmu.2017.01309
Gupta S et al (2013) In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 8:e73957. https://doi.org/10.1371/journal.pone.0073957
Guzman MG et al (2007) Neutralizing antibodies after infection with dengue 1 virus. Emerg Infect Dis 13(2):282–286
Hadinegoro SR, Arredondo-Garcia JL, Capeding MR, Deseda C, Chotpitayasunondh T, Dietze R et al (2015) Efficacy and long-term safety of a dengue vaccine in regions of endemic disease. N Engl J Med 373:1195–1206. https://doi.org/10.1056/NEJMoa1506223
Hasan SS, Sevvana M, Kuhn RJ, Rossmann MG (2018) Structural biology of Zika virus and other flaviviruses. Nat Struct Mol Biol 25:13–20. https://doi.org/10.1038/s41594-017-0010-8
Hope RG, McLauchlan J (2000) Sequence motifs required for lipid droplet association and protein stability are unique to the hepatitis C virus core protein. J Gen Virol 81:1913–1925. https://doi.org/10.1099/0022-1317-81-8-1913
Iglesias NG, Mondotte JA, Byk LA, De Maio FA, Samsa MM, Alvarez C, Gamarnik AV (2015) Dengue virus uses a non-canonical function of the host GBF1-Arf-COPI system for capsid protein accumulation on lipid droplets. Traffic 16:962–977. https://doi.org/10.1111/tra.12305
John AL, Rathore APS (2019) Adaptive immune responses to primary and secondary dengue virus infections. Nat Rev Immunol 19:218–230. https://doi.org/10.1038/s41577-019-0123-x
Kodama Y, Mashima J, Kosuge T, Ogasawara O (2019) DDBJ update: the Genomic Expression Archive (GEA) for functional genomics data. Nucleic Acids Res 47:D69–D73. https://doi.org/10.1093/nar/gky1002
Kuhn RJ et al (2002) Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell 108:717–725. https://doi.org/10.1016/S0092-8674(02)00660-8
London N, Raveh B, Schueler-Furman O (2013) Peptide docking and structure-based characterization of peptide binding: from knowledge to know-how. Curr Opin Struct Biol 23:894–902. https://doi.org/10.1016/j.sbi.2013.07.006
Magalhaes T, Robison A, Young MC et al (2018) Sequential infection of Aedes aegypti mosquitoes with Chikungunya virus and Zika virus enhances early Zika virus transmission. Insects 9(4):E177
Martins IC, Gomes-Neto F, Faustino AF, Carvalho FA, Carneiro FA, Bozz PT, Mohana-Borges R, Castanho MARB, Almeida FCL, Santos NC et al (2012) The disordered N-terminal region of dengue virus capsid protein contains a lipid-droplet-binding motif. Biochem J 444:405–415. https://doi.org/10.1042/BJ20112219
Mathew A et al (2011) B-cell responses during primary and secondary dengue virus infections in humans. J Infect Dis 204(10):1514–1522
Mith-Garvin JE, Koretzky GA, Jordan MS (2009) T cell activation. Annu Rev Immunol 27:591–619. https://doi.org/10.1146/annurev.immunol.021908.132706
Moodie Z et al (2018) Neutralizing antibody correlates analysis of tetravalent dengue vaccine efficacy trials in Asia and Latin America. J Infect Dis 217(5):742–753
Nielsen M, Lund O (2009) NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinform 10:296. https://doi.org/10.1186/1471-2105-10-296
Oliveira ER, De Alencastro RB, Horta BA, Mohana-Borges R (2017) The flavivirus capsid protein: structure, function and perspectives towards drug design. Virus Res 227:115–123. https://doi.org/10.1016/j.virusres.2016.10.005
Osatomi K, Sumiyoshi H (1990) Complete nucleotide sequence of dengue type 3 virus genome RNA. Virology 176:643–647. https://doi.org/10.1016/0042-6822(90)90037-R
Oyarzún P, Kobe B (2016) Recombinant and epitope-based vaccines on the road to the market and implications for vaccine design and production. Hum Vaccine Immunother 12(3):763–767. https://doi.org/10.1080/21645515.2015.1094595
Pandey RK, Ojha R, Mishra A, Kumar PV (2018) Designing B- and T-cell multi-epitope based subunit vaccine using immunoinformatics approach to control Zika virus infection. J Cell Biochem 119(9):7631–7642. https://doi.org/10.1002/jcb.27110
Patkar CG, Jones CT, Chang YH, Warrier R, Kuhn RJ (2007) Functional requirements of the yellow fever virus capsid protein. J Virol 81:6471–6481. https://doi.org/10.1128/JVI.02120-06
Paul D, Bartenschlager R (2015) Flaviviridae replication organelles: Oh, what a tangled web we weave. Annu Rev Virol 2:289–310. https://doi.org/10.1146/annurev-virology-100114-055007
Peters B, Sette A (2007) Integrating epitope data into the emerging web of biomedical knowledge resources. Nat Rev 7(6):485–490. https://doi.org/10.1038/nri2092
Petsalaki E, Russell R B (2008) Peptide-mediated interactions in biological systems: new discoveries and applications. Curr Opin Biotechnol 19:344–350. https://doi.org/10.1016/j.copbio.2008.06.004
Reiter P (2001) Climate change and mosquito-borne disease. Environ Health Perspect 109(Suppl. 1):141–161
Robinson J, Soormally AR, Hayhurst JD, Marsh SGE (2016) The IPD-IMGT/HLA Database—new developments in reporting HLA variation. Hum Immunol 77:233–237. https://doi.org/10.1016/j.humimm.2016.01.020
Rothman AL (2004) Dengue: defining protective versus pathologic immunity. J Clin Investig 113(7):946–951. https://doi.org/10.1172/JCI2151241
Saha S, Raghava GPS (2006) Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 65(1):40–48. https://doi.org/10.1002/prot.21078
Sampath A, Padmanabhan R (2009) Molecular targets for flavivirus drug discovery. Antivir Res 81(1):6–15. https://doi.org/10.1016/j.antiviral.2008.08.004
Samsa MM, Mondotte JA, Iglesias NG, Assunção-Miranda I, Barbosa-Lima G, Da Poian AT, Bozza PT, Gamarnik AV (2009) Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog 5:e1000632. https://doi.org/10.1371/journal.ppat.1000632
Samuel GH, Wiley MR, Badawi A, Adelman ZN, Myles KM (2016) Yellow fever virus capsid protein is a potent suppressor of RNA silencing that binds double-stranded RNA. Proc Natl Acad Sci USA 113:13863–13868. https://doi.org/10.1073/pnas.1600544113
Sathe A, Cusick JK (2020) StatPearls. Biochemistry, Immunoglobulin M Bookshelf ID NBK555995. StatPearls Publishing, Treasure Island
Sayers EW, Cavanaugh M, Clark K, Ostell J, Pruitt KD, Karsch-Mizrachi I (2019) GenBank. Nucleic Acids Res 47:D94–D99. https://doi.org/10.1093/nar/gky989
Servadio JL, Rosenthal SR, Carlson L, Bauer C (2018) Climate patterns and mosquito-borne disease outbreaks in South and Southeast Asia. J Infect Public Health 11(4):566–571. https://doi.org/10.1016/j.jiph.2017.12.006
Shahid F, Ashfaq UA, Javaid A, Khalid H (2020) Immunoinformatics guided rational design of a next generation multi epitope based peptide (MEBP) vaccine by exploring Zika virus proteome. Infect Genet Evol 80:104199. https://doi.org/10.1016/j.meegid.2020.104199
Shang Z, Song H, Shi Y, Qi J, Gao GF (2018) Crystal structure of the capsid protein from Zika virus. J Mol Biol 430:948–962. https://doi.org/10.1016/j.jmb.2018.02.006
Singh H, Raghava GPS (2003) Propred I: prediction of HLA class I binding sites. Bioinformatics 19:1009–1014. https://doi.org/10.1093/bioinformatics/btg108
Slomnicki LP, Chung NH, Parker A, Hermann T, Boyd NL, Hetman M (2017) Ribosomal stress and Tp53-mediated neuronal apoptosis in response to capsid protein of the Zika virus. Sci Rep 7:16652. https://doi.org/10.1038/s41598-017-16952-8
Soto-Acosta R, Bautista-Carbajal P, Syed GH, Siddiqui A, Del Angel RM (2014) Nordihydroguaiaretic acid (NDGA) inhibits replication and viral morphogenesis of dengue virus. Antivir Res 109:132–140. https://doi.org/10.1016/j.antiviral.2014.07.002
Stadler MB, Stadler BM (2003) Allergenicity prediction by protein sequence. FASEB J 17:1141–1143. https://doi.org/10.1096/fj.02-1052fje
Subramaniyan V, Venkatachalam R, Srinivasan P, Palani M (2017) In silico prediction of monovalent and chimeric tetravalent vaccines for prevention and treatment of dengue fever. J Biomed Res 32(3):222–236. https://doi.org/10.7555/JBR.31.20160109
Tahir ul Qamar M, Bari A, Adeel MM et al (2018) Peptide vaccine against Chikungunya virus: immuno-informatics combined with molecular docking approach. J Transl Med 16:298. https://doi.org/10.1186/s12967-018-1672-7
Tanaka S, Baba Y (2020) B cell receptor signaling. Adv Exp Med Biol 1254:23–36
Thiele C, Spandl J (2008) Cell biology of lipid droplets. Curr Opin Cell Biol 20:378–385. https://doi.org/10.1016/j.ceb.2008.05.009
Thomas SJ, Yoon IK (2019) A review of Dengvaxia: development to deployment. Hum Vaccine Immunother 15(10):2295–2314
Tong JC, Ren EC (2009) Immunoinformatics: current trends and future directions. Drug Discov Today 14(13–14):684–689. https://doi.org/10.1016/j.drudis.2009.04.001
Varjak M, Donald CL, Mottram TJ, Sreenu VB, Merits A, Maringer K, Schnettler E, Kohl A (2017) Characterization of the Zika virus induced small RNA response in Aedes aegypti cells. PLoS Negl Trop Dis 11:e0006010. https://doi.org/10.1371/journal.pntd.0006010
Verma M, Bhatnagar S, Kumari K, Mittal N, Sukhralia S, Gopirajan At S, Dhanaraj PS, Lal R (2019) Highly conserved epitopes of DENV structural and non-structural proteins: candidates for universal vaccine targets. Gene 695:18–25. https://doi.org/10.1016/j.gene.2019.02.001
Villar L, Dayan GH, Arredondo-García JL, Rivera DM, Cunha R, Deseda C, Reynales H, Costa MS, Morales-Ramírez JO, Carrasquilla G et al (2015) Efficacy of a tetravalent dengue vaccine in children in Latin America. N Engl J Med 372:113–123. https://doi.org/10.1056/NEJMoa1411037
Vita R, Zarebski L, Greenbaum JA, Emami H, Hoof I, Salimi N, Damle R, Sette A, Peters B (2010) The immune epitope database 2.0. Nucleic Acids Res 38(Database):D854-862. https://doi.org/10.1093/nar/gkp1004
Vogels CBF, Rückert C, Cavany SM, Perkins TA, Ebel GD, Grubaugh ND (2019) Arbovirus coinfection and co-transmission: a neglected public health concern? PLoS Biol 17(1):e3000130
Wahala WMPB (2011) de Silva IS (2011) The human antibody response to dengue virus infection. Viruses 3(12):2374–2395. https://doi.org/10.3390/v3122374 (Epub 25 Nov 2011)
Zellweger RM, Eddy WE, Tang WW, Miller R, Shresta S (2014) CD8+ T cells prevent antigen-induced antibody-dependent enhancement of dengue disease in mice. J Immunol 193:4117–4124. https://doi.org/10.4049/jimmunol.1401597
Zhu W, Qin C, Chen S, Jiang T, Yu M et al (2007) Attenuated dengue 2 viruses with deletions in capsid protein derived from an infectious full-length cDNA clone. Virus Res 126:226–232. https://doi.org/10.1016/j.virusres.2007.03.004
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Dixit, N.K. Design of Monovalent and Chimeric Tetravalent Dengue Vaccine Using an Immunoinformatics Approach. Int J Pept Res Ther 27, 2607–2624 (2021). https://doi.org/10.1007/s10989-021-10277-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10989-021-10277-x