
Abstract
Radiolabeled fatty acids as myocardial metabolic agent are used for detecting ischemic heart disease, however, no 99mTc-labeled fatty acids have potential use in clinical diagnosis. In this work, five fatty acid analogues labeled with 99mTc were firstly synthesized and characterized, their biological behaviors were investigated. All Radiotracers had good stability when incubated in rat serum for 3 h at 37 °C. 99mTc -CpT-12-ODPPA (8b) showed higher initial myocardial uptake (8.17% ID/g at 1 min postinjection) and good heart/blood ratio (2.58 at 30 min postinjection). 99mTc-11-dpa-OUFA (2b) as positively charged lipophilic compounds had reasonable heart uptake (5.59% ID/g at 1 min postinjection) and good retention (1.89% ID/g at 60 min postinjection). Unfortunately, no great improvement was obtained by the five 99mTc-labeled fatty acid analogues for the short myocardial retention and poor heart/liver ratios.
Similar content being viewed by others


Introduction
Worldwide, heart diseases cause death and deformity, which not only pose a threat to human health but also bring a heavy burden to society in terms of economic and social costs. Early diagnosis of cardiovascular diseases is the key to reduce the mortality of cardiovascular diseases, and the research and development of new type of diagnostic radiopharmaceuticals is very important for early diagnosis and treatment of cardiovascular diseases. Myocardial metabolism imaging can characterize the viability of myocardium, myocardial metabolic function and blood flow. The viability of myocardium which is the most important evaluation index plays a major role in diagnosis of cardiovascular diseases [1].
Free long chain fatty acids provide energy for myocardium, and it can contribute about 60–80% of ATP (adenosine triphosphates) in normal myocardium by β-oxidation under aerobic or low workload condition [2, 3]. The metabolic fate of free long chain fatty acids depends on blood flow, oxygen supply, concentration, the cardiac workload and the presence or absence of ischemia [1, 4,5,6]. When the human or animals are in the fasted state, long chain fatty acids provide most of the ATP required by the heart; however, the availability of fatty acids is suppressed and glucose acts as an energy source in stunning and full state [7]. Thus, fatty acid uptakes are considered to be a sensitive signal of ischemia and myocardial damage, and the long chain fatty acids labelled by radionuclide might be of great importance in myocardial imaging [8, 9].
In recent years, numerous studies in radio-labeled fatty acids have been carried out and some successes have been achieved. [123I]IPPA, [123I]BMIPP and [11C]palmitic acid have been extensively used in clinic. However, due to the short half-life of 11C (t 1/2 = 20 min) and rapid biexponential clearance of [11C]palmitic acid from myocardium, the utilization of [11C]palmitic acid has been gravely restricted [10,11,12]. As for two types of iodine labelled compounds, they are easy to shed radioiodine, which has affected the widespread use of them in radiopharmaceuticals. Though 99mTc possesses desirable characteristics (Eγ = 141 keV, t 1/2 = 6.02 h, easily obtained through the 99Mo/99mTc generator system), none of 99mTc-labelled fatty acid analogues have been applied in clinic [13, 14], so it is very challenging to develop a potential 99mTc-labelled fatty acid metabolic imaging agent.
In 2008, 99mTc-CpTT-16-oxo-HAD (Fig. 1) was studied and the results showed that it had high uptake in heart through β-oxidation in myocardium [15]. Zeng et al. [16] introduced the thiophene ring at the β-position of the long chain fatty acid (Fig. 1) in 2014, and the results showed that the 99mTc-CpTT-15-oxo-PTA has higher initial myocardial uptake (9.39% ID/g) and higher heart/blood ratio (5.7 at 15 min postinjection), which suggested further study of those compounds is worthwhile.

The structure of 99mTc-CpTT-16-oxo-HDA and 99mTc-CpTT-15-oxo-PTA
In 2008 [17, 18], Mathur et al. reported FA-cysteine conjugated with [99mTcPNP]2+ core and subsequently synthesized its positively charged structural analogue, and showed the positively charged fatty acid derivatives seems to be better. Cazzola et al. [19] also reported a series of neutral and positively charged fatty acid derivatives labeled with [99mTcN(PNP)]2+ and found only the monocationic complexes had signification heart uptake, the corresponding neutral compound had some absorption only in the former 5 min, but completely washed out after 30 min.
In the present study, we designed and synthesized two novel fatty acid analogues, and some biological evaluations were conducted. The envisaged strategy involved the synthesis and bio-evaluation of three fatty acid derivatives with dipicolylamine ligand and incorporated with fac-[99mTc(CO)3]+ (Scheme 1). They were monocationic complexes and we inserted furan ring at the C4 position. The initial aspiration was to increase the heart uptake and to reduce the uptake of non-targeted organs.

The synthesis of compound 4, 5 and 6
Another, we replaced the thiophene ring with its isotere pyrrole ring or introduced amidate group (Scheme 2), because of the stability and smaller molecular size of cyclopentadienyl tricarbonyl technetium-99m may be beneficial to myocardial accumulation.

The synthesis of compound 7, 8, 9
Experimental section
Materials
Chemicals and solvents were obtained from commercial suppliers and used without further purification. Na99mTcO4 was obtained by elution with saline from a 99Mo-99mTc generator (Beijing Atomic High-tech Co., Beijing). Tissue radioactivity in an automatic γ-Counter. NMR spectra were recorded on a 400-MHz spectrometer (Bruker), A Trance MS-2000 mass spectrometer (Finnigan) was used to obtain Mass spectra (EI-MS) spectra. Reversed-phase high-performed liquid chromatography (HPLC) was conducted on a system equipped with an LC-20AT pump (Shimadzu), a B-FC-320 flow counter (Bioscan) and UV (254 nm). The vitro cytotoxicity assay was examined at 570 (for absorbance of MTT formazan) and 650 nm (for the reference wavelength) in a microplate reader (BioTek Instruments Inc, USA). All animal experiments were carried out by using normal mice (Vital River Laboratory Animal Technology Co. Ltd., China, Yangshan Road No. 4, Chaoyang district, Beijing), and in compliance with the relevant national laws, as approved by the local committee on the conduct and ethics of animal experimentation.
Synthesis
Synthesis of ethyl 3-(5-(12-bromododecanoyl) furan-2-yl) propanoate (1)
A dry 250 mL round-bottom flash charged with 2.78 g (0.01 mmol) 12-bromododecanoic acid, anhydrous CH2Cl2 (40 mL), 2 drops of DMF and oxalyl chloride (1.5 mL), the solution was stirred at room temperature for 3 h. After evaporating the CH2Cl2 and the excess oxalyl chloride, dry CH2Cl2 (15 mL) and ethyl 3-(furan-2-yl) propanoate (1.68 g, 0.01 mmol) dissolved in dry CH2Cl2 (10 mL) was added dropwise in an ice bath. The mixture was stirred for 10 min, then adding anhydrous AlCl3 (1.34 g, 0.01 mmol) carefully. After stirred for 30 min, the crude mixture was diluted with a solution of 1 M hydrochloric acid, extracted with CH2Cl2 (3 × 50 mL) and dried over anhydrous Na2SO4. The residue was subjected to silica gel chromatography (PE/EA = 12:1–8:1, v/v) to obtained 1 (3.23 g, 75.5%) as yellow oil. 1H-NMR (CDCl3, 400 MHz) δ 7.08 (d, J = 3.4 Hz, 1H, furan(β)), 6.19 (d, J = 3.4 Hz, 1H, furan(β)), 4.12–4.17 (m, 2H), 3.40 (t, J = 6.9 Hz, 2H), 3.04 (t, J = 7.4 Hz, 2H), 2.68–2.76 (m, 4H), 1.81–1.88 (m, 2H), 1.70–1.72 (m, 2H), 1.59–1.65 (m, 2H), 1.39–1.43 (m, 17H).
Synthesis of ethyl 3-(5-(12-(bis(pyridin-2-ylmethyl)amino)dodecanoyl)furan-2-yl) propanoate (4)
The method reported in the previous literature [20]. Bis(pyridin-2-ylmethyl)amine (843.75 mg, 4.2 mmol), diisopropylethylamine (DIEA) (1.5 mL, 8.4 mmol), KI (69 mg, 0.42 mmol), and compound 1 (1.54 g, 3.6 mmol) were dissolved in acetonitrile (40 mL), the solution was stirred at 70 °C for 2 h under the protection of N2. The temperature heated up to 85 °C, the mixture was refluxed overnight. The crude product was washed with saturated salt water (5 × 30 mL), extracted with CH2Cl2 and dried over anhydrous Na2SO4. Removed excess acetonitrile on a rotary evaporator, the residue was subjected to silica gel chromatography (PE/EA = 8:3, containing 1% v/v TEA) to obtained compound 4 as yellow oil (1.5 g, yield 76.1%). 1H-NMR (CDCl3, 400 MHz) δ 8.48–8.50 (m, 2H, –pyridine), 7.61–7.75 (m, 2H, –pyridine), 7.54 (d, J = 7.7 Hz, 2H, –pyridine), 7.06–7.13 (m, 2H, –pyridine), 7.06 (d, J = 3.4 Hz, 1H, –furan), 6.18 (d, J = 3.4 Hz, 1H, –furan), 4.15 (q, J = 7.2 Hz, 2H 2H, –O–CH2–), 3.79 (s, 4H, –N–CH2–), 3.02 (t, J = 7.4 Hz, 2H, –furan–CH 2–CH2–), 2.66–2.73 (m, 4H), 2.51 (t, J = 7.4 Hz, 2H, –C = O–CH 2–), 1.64–1.68 (m, 2H), 1.49–1.51 (m, 2H), 1.19–1.24 (m, 22H); 13C-NMR (CDCl3, 100 MHz) δ 189.17 (–furan–C=O), 171.88 (–COO–), 159.18 (–furan), 151.68(–furan), 148.87 (–pyridine), 136.31(–pyridine), 122.87(–pyridine), 121.85(–pyridine), 118.40 (–furan), 108.44 (–furan), 60.65 (–O–CH2–), 60.34 (–N–CH2–), 54.36, 38.21, 32.06, 29.47, 29.35, 29.28, 27.20, 26.89, 24.52, 23.68, 14.14; MS (ESI) m/z: calcd for [C33H45N3O4] 547.8; found: (M+H)+ 548.4.
Synthesis of ethyl 3-(5-(11-(bis (pyridin-2-ylmethyl) amino) undecanoyl) furan-2-yl) propanoate (5)
The synthesis method of compound 5 is similar to that of 4. 1H-NMR (CDCl3, 400 MHz) δ 8.50–8.51 (m, 2H, –pyridine), 7.62–7.64 (m, 2H, –pyridine), 7.55 (d, J = 8.0 Hz, 2H, –pyridine), 7.13 (t, J = 5.2 Hz, 2H, –pyridine), 7.07 (d, J = 3.6 Hz, 1H, –furan), 6.18 (d, J = 3.2 Hz, 1H, –furan), 4.15 (q, J = 7.2 Hz, 2H, –O–CH2–), 3.81 (s, 4H, –N–CH2–), 3.05 (t, J = 7.2 Hz, 2H, –furan–CH 2–CH2–), 2.74–2.67 (m, 4H), 2.51 (t, J = 7.0 Hz, 2H, –C=O–CH 2–), 1.67 (m, 2H), 1.25–1.28 (m, 2H), 1.20–1.24 (m, 17H); 13C-NMR (CDCl3, 100 MHz) δ 189.15 (–furan–C=O), 171.88 (–COO–), 159.99 (–furan), 151.77 (–furan), 148.77 (–pyridine), 136.34 (–pyridine), 122.81 (–pyridine), 121.81 (–pyridine), 118.40 (–furan), 108.45 (–furan), 60.64 (–O–CH2–), 60.37 (–N–CH2–), 54.41, 38.19, 32.06, 29.44, 29.34, 29.25, 27.65, 27.21, 26.98, 24.50, 23.68, 19.10, 14.13; MS (ESI) m/z: calcd for [C32H43N3O4] 533.7; found: (M+H) + 534.4.
Synthesis of ethyl 3-(5-(10-(bis(pyridin-2-ylmethyl)amino)decanoyl)furan-2-yl) Propanoate (6)
The synthesis method of compound 6 is similar to that of 4. 1H-NMR (CDCl3, 400 MHz) δ 8.48–8.49 (m, 2H, –pyridine), 7.61–7.62 (m, 2H, –pyridine), 7.54 (d, J = 7.0 Hz, 2H, –pyridine), 7.11 (t, J = 7.0 Hz, 2H, –pyridine), 7.05 (s, 1H, –furan), 6.17 (s, 1H, –furan), 4.13 (q, J = 7.0 Hz, 2H, –O–CH2–), 3.79 (s, 4H, –N–CH2–), 3.01 (t, J = 7.0 Hz, 2H, –furan–CH 2–CH2–), 2.68 (t, J = 7.6 Hz, 2H), 2.50 (s, 2H, –C=O–CH 2–), 1.56–1.64 (m, 2H), 1.50–1.52 (m, 2H), 1.20–1.26 (m, 15H); 13C-NMR (CDCl3, 100 MHz) δ 189.19 (–furan–C=O), 171.93 (–COO–), 159.23 (–furan), 151.73 (–furan), 148.91 (–pyridine), 136.36 (–pyridine), 122.91 (–pyridine), 121.89 (–pyridine), 118.45 (–furan), 108.49 (–furan), 60.70 (–O–CH2–), 60.41 (–N–CH2–), 54.40, 38.24, 32.11, 29.35, 29.29, 27.22, 26.96, 24.53, 23.73, 14.19; MS (ESI) m/z: calcd for [C31H41N3O4] 519.72; found: (M+H)+ 520.3917.
Synthesis of Methyl 3-(3-(thiophen-2-yl)propanamido)propanoate (7)
Formic acid (50 mL) was added to triethylamine (142 mL) dropwise in ice bath along with gas, then heated for 6 h at 60 °C. After cooled to room temperature, the mixture was extracted with ether to obtain colorless liquid (TEAF). 2-Thenaldehyde (56.1 g, 0.5 mol) and 2,2-Dimethyl-1,3-dioxane-4,6-dione (79.3 g, 0.5 mol) were dissolved into TEAF (200 mL). After refluxing at 95 °C for 6 h, the mixture was extracted with dichloromethane. Removing the solvent, oily liquid was 2-Thiophenepropanoic acid. Added Thionyl chloride (20 mL) and dichloromethane (100 mL) to 2-Thiophenepropanoic acid (27 g, 0.17 mol), the mixture reacted 6 h at room temperature. When the solvent was evaporated, 3-(thiophen-2-yl)propanoyl chloride was given [21, 22]. Methyl 3-aminopropionate hydrochloride (16.8 g, 0.12 mol) was dissolved in dry DMF (150 mL), then added with triethylamine (18 g, 0.18 mol). Stirred for 10 min, 3-(thiophen-2-yl)propanoyl chloride (20.6 g, 0.12 mol) in 50 mL dry DMF was added slowly in ice bath and stirred for 4 h. The reaction mixture was poured into water (15 mL) and extracted with dichloromethane. When the solvent was evaporated, the residue was subjected to chromatography (1:4–1:2, v/v, ethyl acetate-petroleum ether) to give 7 [23] (10.5 g, 36.3%). 1H-NMR (CDCl3, 400 MHz) δ 7.10–7.12 (dd, J 1 = 5.1 Hz, J 2 = 1.1 Hz, 1H, thiophene), 6.88–6.91 (dd, J 1 = 5.1 Hz, J 2 = 3.4 Hz, 1H, thiophene), 6.80–6.81 (dd, J 1 = 3.3 Hz, J 2 = 0.8 Hz, 1H, thiophene), 6.02 (s, 0.8H, NH), 3.68 (s, 3H), 3.46–3.51 (q, J = 6.1 Hz, 2H), 3.17 (t, J = 7.4 Hz, 2H), 2.48–2.52 (m, 4H). MS (ESI) m/z: calcd for [C11H14NO3S] 241.1; found (Pos): (M+H)+ 242.1, (M+Na)+ 264.1.
Synthesis of methyl 12-ferrocene-12-oxo-dodecanoyl pyrrolpropanoate (8)
The method reported in the previous literature [24]. A mixture of thionyl chloride (15 mL, 206 mmol) and dodecanedioic acid (3.15 g, 11 mmol) was heated at 90 °C for 4 h. After removed excess thionyl chloride, the 1, 12-dodecanedioyl dichlorides was obtained and used without further purification. Ferrocene (2.16 g, 11 mmol) and 1,12-dodecanedioyl dichloride in dichloromethane (30 mL) was stirred in ice bath, then anhydrous AlCl3 (1.5 g, 11.2 mmol) was added carefully. After the mixture was reacted for 1 h at room temperature, the methyl 3-(1-H-pyrrol-2-yl) propanoate (2.61 g, 16.5 mmol) dissolved in 15 mL CH2Cl2 was added in an ice bath. After 30 min, anhydrous AlCl3 (1.5 g, 11.2 mmol) was added carefully. After reacted for 1 h, the crude mixture was diluted with 1 M HCl. The organic layer was extracted with CH2Cl2 and dried over anhydrous Na2SO4. When the excess solvent was removed, the residue was purified by chromatography (1:10–1:4, v/v, ethyl acetate-petroleum ether) to give 8 (640 mg, 10.9%). 1H-NMR (CDCl3, 400 MHz) δ 9.39 (s, 1H, –NH), 6.79 (t, J = 2.6 Hz, 1H, pyrrol(α)), 5.99 (t, J = 2.6 Hz, 1H, pyrrol(β)), 4.78 (t, J = 1.9 Hz, 2H, -Fe-Cp-H(α)), 4.48 (t, J = 1.89 Hz, 2H, –Fe–Cp–H(β)), 4.19 (s, 5H, Cp–H), 3.70 (s, 3H, –O–CH3), 2.94 (t, J = 7.1 Hz, 2H), 2.64–2.71 (m, 4H), 1.68–1.71 (m, 4H), 1.30 (s, 12H), 13C-NMR (CDCl3, 100 MHz) δ 204.74 (–Fe–Cp–C=O), 190.53 (–pyrrol–C=O), 173.31 (–COO–), 138.12 (–pyrrol), 131.53(–pyrrol), 116.60 (–pyrrol), 108.63 (–pyrrol), 79.34 (–Fe–C–C=O), 72.13 (–Cp), 69.80 (–Cp), 69.42 (–Cp), 52.01 (–O–CH3), 39.86, 37.79, 33.56, 29.77, 29.63, 29.54, 29.49, 25.50, 24.72, 22.92. MS (ESI) m/z: calcd for [C30H39NO4Fe] 533.5; found (Pos): (M+H)+ 534.3, (M+Na) + 556.5.
Synthesis of Methyl 12-ferrocene-12-oxo-dodecanoyl 3-(3-(thiophen-2-yl)propanamido) propanoate (9)
Synthetic procedure was similar to 8. The residue was subjected to chromatography (1:2–1:1, v/v, ethyl acetate-petroleum ether) to give 9 (500 mg, 4.9%). 1H-NMR (CDCl3, 400 MHz) δ 7.52 (s, 1H, thiophene (α)), 6.85 (s, 1H, thiophene (β)), 6.08 (s, 1H, –NH), 4.77 (s, 2H, –Fe–Cp–H(α)), 4.48 (s, 2H, –Fe–Cp–H(β)), 4.19 (s, 5H, Cp–H), 3.68 (s, 3H, –O–CH3), 3.50 (q, J = 5.6 Hz, 2H, –NH–CH 2–), 3.18 (t, J = 7.3 Hz, 2H), 2.81 (t, J = 7.4 Hz, 2H), 2.68 (t, J = 7.4 Hz, 2H), 2.49–2.54 (m, 4H), 1.65–1.73 (m, 4H), 1.22–1.30 (s, 12H); 13C-NMR (CDCl3, 100 MHz) δ 203.87 (–Fe–Cp–C=O), 192.32 (–pyrrol–C=O), 171.95 (–COO–), 170.07 (–CONH–), 151.86 (thiophene), 141.47 (thiophene), 131.09 (thiophene), 125.25 (thiophene), 78.16 (–Fe–C–C=O), 71.13 (–Cp), 68.73 (–Cp), 68.34 (–Cp), 50.80 (–O–CH3), 38.74 (–NH–CH2–), 37.99, 36.66, 33.93, 32.76, 28.51, 28.45, 28.40, 28.35, 28.31, 28.18, 28.09, 25.35, 23.88, 23.64. MS (ESI) m/z: calcd for [C33H43NO5SFe] 621.2; found (Pos): (M+H+) 622.1, (M+Na+) 644.3.
Synthesis of tricarbonyl-Re-(bis((pyridine-2-yl)-methyl)amino)-12-oxo-dodecanoyl furanpropinic acid (Re-12-dpa-ODFA, 1a)
[Re(CO)3(H2O)3]Br was prepared using the method reported in the previous literature [25]. Bromopentacar-bonylrhenium 200 mg (0.48 mmol) was dissolved in excess water (10 mL). After refluxed for 24 h, the water was removed, [Re(CO)3(H2O)3]Br was obtained as a light green powder. Without future processing, [Re(CO)3(H2O)3]Br dissolved into water (5 mL) and compound 4 (180 mg) dissolved into CH3CN (2 mL) was mixed, then stirred for 2 h at 100 °C, the reaction was monitored using TLC. After the reaction was completed, NaOH (2 mL, 1 N) was directly added and heated at 100 °C for 1 h, then acidification with hydrochloric acid (1 M) to pH 7, the organic phase was extracted with dichloromethane and dried over Na2SO4. The residue was purified by column chromatography (CH2Cl2:CH3OH = 50:1, containing 1% glacial acetic acid). 1H-NMR (CDCl3, 400 MHz) δ 9.31 (s, –COOH), 8.64 (d, J = 5.2 Hz, 2H, –pyridine), 7.88 (s, 2H, –pyridine), 7.82 (t, J = 7.3 Hz, 2H, –pyridine), 7.20 (t, J = 6.3 Hz 2H, –pyridine), 7.09 (d, J = 3.2 Hz, 1H, –furan), 6.22 (d, J = 3.1 Hz, 1H, –furan), 5.50 (d, J = 16.7 Hz, 2H, –O–CH2–), 4.47 (d, J = 16.5 Hz, 2H, –N–CH2–), 3.67 (t, J = 7.4 Hz, 2H, –furan–CH 2–CH2–), 3.01 (t, J = 7.3 Hz, 2H, –furan–CH2–CH 2–), 2.73 (t, J = 7.4 Hz, 2H, –C=O–CH 2–), 2.66 (t, J = 7.4 Hz, 2H, –C=O–CH2–CH 2–), 1.67–1.72 (m, 2H), 1.29–1.36 (m, 16H); 13C-NMR (CDCl3, 100 MHz) δ 195.94 (–Re–C=O), 190.01 (–furan–C=O), 176.11 (–COO–), 161.61 (–pyridine), 161.55 (–pyridine), 160.82 (–pyridine), 151.69 (–furan), 150.60 (–furan), 140.37 (–pyridine), 130.19 (–pyridine), 125.17 (–pyridine), 125.07 (–pyridine), 125.00 (–pyridine), 136.31 (–pyridine), 118.58 (–furan), 108.37 (–furan), 71.18 (–N–CH2–), 67.39 (–N–CH2–), 38.33, 29.70, 29.32, 29.18, 28.79, 28.65, 28.53, 28.31, 26.88, 25.34, 25.16, 24.98, 24.90, 24.72, 22.17; MS (ESI) m/z: calcd for [C34H 18740 ReN3O7] 789.74; found: (M+) 790.3178, (M+H+) 791.3215.
Synthesis of tricarbonyl-Re-(bis((pyridine-2-yl)-methyl)amino)-11-oxo-undecanoyl furanpropinic acid (Re-11-dpa-OUFA,2a)
The synthesis method of compound 2a is similar to that of 1a. 1H-NMR (CDCl3, 400 MHz) δ 8.64 (d, J = 5.2 Hz, 2H, –pyridine), 7.97 (t, J = 7.0 Hz, 2H, –pyridine), 7.81 (t, J = 7.3 Hz, 2H, –pyridine), 7.19 (t, J = 6.6 Hz, 2H, –pyridine), 7.11 (d, J = 3.2, 1H, –furan), 6.23 (s, 1H, –furan), 5.72 (d, J = 17.0 Hz, 2H, –O–CH2–), 4.43 (d, J = 16.5 Hz, 2H, –N–CH2–), 3.68 (t, J = 8.9 Hz, 2H, –furan–CH 2–CH2–), 3.08 (t, J = 8.7 Hz, 2H, –furan–CH2–CH 2–), 2.78 (t, J = 7.6 Hz, 2H, –C=O–CH 2–), 2.72 (t, J = 7.9 Hz, 2H, –C=O–CH2–CH 2–), 2.20–2.23 (m, 2H), 1.75 (m, 2H), 1.59–1.66 (m, 2H), 1.32–1.34 (m, 14H); 13C-NMR (CDCl3, 100 MHz) δ 195.97 (–Re–C=O), 189.86 (–furan–C=O), 176.69 (–COO–), 161.30 (–pyridine), 160.31 (–pyridine), 150.68 (–furan), 140.37 (–furan), 129.90 (–pyridine), 125.28 (–pyridine), 125.13 (–pyridine), 125.07 (–pyridine), 118.49 (–furan), 108.48 (–furan), 71.26 (–N–CH2–), 62.37(–N–CH2–), 45.52, 38.15, 31.91, 29.69, 29.51, 29.35, 29.31, 29.21, 29.10, 29.01, 28.86, 28.48, 22.68; MS (ESI) m/z: calcd for [C33H 18738 ReN3O7] 775.71; found: (M+) 776.2. (M+H+) 767.6.
Synthesis of tricarbonyl-Re-10-(bis((pyridine-2-yl)-methyl)amino)-10-oxo-dcanoyl furanpropinic acid (Re-10-dpa-ODFA, 3a)
The synthesis method of compound 3a is similar to that of 1a. 1H-NMR (CDCl3, 400 MHz) δ 8.66 (d, J = 7.6 Hz, 2H, –pyridine), 7.89 (t, J = 7.6 Hz, 2H, –pyridine), 7.83 (t, J = 7.4 Hz, 2H, –pyridine), 7.62 (s, COOH), 7.24 (t, J = 6.2 Hz, 2H, –pyridine), 7.08 (d, J = 3.3 Hz, 1H, –pyridine), 6.21 (d, J = 3.0 Hz, 1H, –pyridine), 5.70 (d, J = 16.7 Hz, 2H, –O–CH2–), 4.42 (d, J = 16.5 Hz, 2H, –N–CH2–), 3.70 (t, J = 6.0 Hz, 2H, -furan–CH 2–CH2–), 2.68–2.75 (m, 4H), 2.01 (t, J = 6.7 Hz, 2H), 1.67–1.69 (m, 2H), 1.33–1.37 (m, 12H); 13C-NMR (CDCl3, 100 MHz) δ 195.91 (–Re–C=O), 189.66 (–furan–C=O), 176.13 (–COO–), 161.00 (–-pyridine), 160.34 (–pyridine), 150.82 (–furan), 140.40 (–furan), 129.90 (–pyridine), 125.43 (–pyridine), 125.13 (–pyridine), 124.93 (–pyridine), 118.88 (–furan), 108.52 (–furan), 71.11 (–N–CH2–), 67.32 (–N–CH2–), 45.44, 38.10, 29.68, 29.30, 29.07, 29.02, 28.96, 26.79, 25.53, 24.76, 21.62; MS (ESI) m/z: calcd for [C32H 18736 ReN3O7] 760.68; found: (M+) 762.34. (M+H+) 763.34.
Synthesis of 12-cyclopentadienyl tricarbonyl rhenium 12-oxo-dodecanoy pyrrolpropinic acid (Re-CpT-12-ODPPA, 8a)
The method reported in the previous literature [26]. A high-pressure tanks of polytetrafluoroethylene containing ester 8 (200 mg), NH4ReO4 (60 mg, 210 μmol), Cr(CO)6 (255 mg, 1155 mmol), CrCl3 (60 mg, 0.39 mmol) and methanol (900 μL) was heated at 180 °C oil bath for 2 h. At the completion of the reaction, the mixture was cooled and extracted with CH2Cl2. After solvent was removed under vacuum, methanol (3 mL) and 0.3 M NaOH (1 mL) heated at 80 °C for 40 min. The pH value of solution was acidified to 7 with 0.1 M HCl. The reaction mixture was concentrated under reduced pressure and the crude products were purified by flash column chromatography (1:2 ethyl acetate: petroleum ether) to give the 8a (18 mg, 7.2%). 1H-NMR (CDCl3, 400 MHz) δ 10.60 (s, 1H, –NH), 6.89 (s, 1H, pyrrol(α)), 6.04 (s, 1H, pyrrol(β)), 5.98 (s, 2H, –Re–Cp–H(α)), 5.39 (s, 2H, –Re–Cp–H(β)), 3.01 (t, J = 7.1 Hz, 2H), 2.68–2.76 (m, 4H), 2.57 (t, J = 7.2 Hz, 2H), 1.26 (s, 16H); 13C-NMR (CDCl3, 100 MHz) δ 195.42 (–Re–C=O), 191.84 (–COO–), 176.09 (–pyrrol–C=O), 140.17 (–pyrrol–), 118.80 (–pyrrol–), 109.03 (–pyrrol–), 96.20 (–Re–C–C=O), 87.91 (–Re–Cp(α)), 85.12 (–Re–Cp(β)), 38.85, 37.80, 33.57, 29.41, 29.32, 29.28, 29.05, 25.65, 24.41, 22.79. MS (ESI) m/z: found (Neg): (M-H+, 185 Re) 666.2, (M-H+, 187 Re) 668.1.
Synthesis of 12-cyclopentadienyl tricarbonyl rhenium 12-oxo-dodecanoy 3-(3-(thiophen-2-yl)propanamido)propanoate acid (Re-CpT-12- ODTPPA, 9a)
Compound 9a (4 mg, 1.6%) was obtained as described in the procedure of 8a. 1H-NMR (CDCl3, 400 MHz) δ 7.52 (d, J = 3.7 Hz, 1H, thiophene (α)), 6.85 (d, J = 3.7 Hz, 1H, thiophene (β)), 6.04 (s, 0.8H, –NH), 5.98 (t, J = 2.2 Hz, 2H, –Re–Cp–H(α)), 5.39 (t, J = 2.2 Hz, 2H, –Re–Cp–H(β)), 3.52 (q, J = 5.9 Hz, 2H), 3.19 (t, J = 7.4 Hz, 2H), 2.82 (t, J = 7.4 Hz, 2H), 2.58 (t, J = 7.3 Hz, 2H), 2.49–2.54 (m, 4H), 1.61–1.75 (m, 4H), 1.25–1.28 (s, 12H); 13C-NMR (CDCl3, 100 MHz) δ 194.59 (–Re–C=O), 192.28 (–Re–C=O), 190.86 (–Re–C=O), 177.15 (–COO–), 176.53 (–thiophene-C=O), 150.91 (–thiophene), 139.94 (–thiophene), 130.74 (–thiophene), 123.97 (–thiophene), 95.14 (–Re–C–C=O), 86.95 (–Re–Cp(α)), 84.18 (–Re–Cp(β)), 37.99 (–NH–CH2–), 37.82, 35.09, 31.81, 28.68, 28.27, 28.23, 28.20, 28.02, 26.20, 25.09, 23.90, 23.39; MS (ESI) m/z: found (Pos): (M+H+, 187 Re) 772.1, (M+Na+, 187 Re) 794.1.
Radiochemical synthesis
The labeling procedure of 8b and 9b, as shown in Scheme 3, the method reported in the previous literature [27]. Ester 8, 9 (1.5 mg), Mn(CO)5Br (4.5 mg) and DMF (1.0 mL) were placed in the sealed vial respectively, Na99mTcO4 solution (25 mCi) was added and then heated at 150 °C (oil bath) for 1 h. At the end of the reaction, cooled the mixture rapidly in an ice bath and dissolved the residue in dichloromethane, passed it through a 0.22 μm Millipore filter, then removed the solvent. The residue was dissolved in methanol (300 μL), then added 0.3 M NaOH (100 μL) at 80 °C for 15 min. Cooled, acidified with 0.1 M HCl (150 μL) and extracted with dichloromethane.

Synthetic procedure for radio-tracers and Re complexes. Reaction conditions: a Re(CO)3(H2O)3Br, 100 °C, 2 h; b 0.3 N NaOH-CH3OH(1:3) 80 °C; c [99mTc(H2O)3(CO)3]+, 80 °C, pH 8; d 0.3 N NaOH-CH3OH(1:3), 80 °C, 10 min; e NH4ReO4, Cr(CO)6, CrCl3, CH3OH, 180 °C, 1 h; f Na99mTcO4, Mn(CO)5Br, DMF, 150 °C, 1 h
The labeling procedure of 1b, 2b and 3b, as shown in Scheme 3, the synthesis of intermediate was done on the basis of the literature reported by Alberto [28]. Na2CO3 (5 mg), NaBH4 (10 mg), potassium sodium tartrate (15 mg) and saline (1 mL) in a 10 mL glass vial, then the vial was sealed. After purging CO gas to the solution for 15 min, Na99mTcO4 (10 mCi) was added, then the solution was heated at 80 °C for 30 min. The chemical purity of the precursor was over 90% characterized by HPLC. Prepared intermediate fac-[99mTc(CO)3(H2O)3]+ (0.5 mL, 5 mCi) was added to 4, 5 or 6 (0.3 mg dissolved in 300 μL saline), then adding 1 M HCl to adjust the pH 8, the mixture was reacted at 80 °C for 30 min. The next step was adding NaOH (0.5 mL, 1 mol/L) and reacted for 20 min. The reaction solution was cooled to room temperature and acidified to pH 7 with 1 mol/L HCl and extracted with CH2Cl2.
After removing the solvent under the N2, the crude product was purified by radio-HPLC after passing through 0.22 μm Millipore filter. The retention time of the five radiotracers (1b, 2b, 3b, 8b and 9b) were observed to be according to the corresponding Re counterparts (Fig. 2). The final radiochemical purity was analyzed by re-injecting the product onto the radio-HPLC.

HPLC co-elution profiles of 1a.1b, 2a.2b, 3a.3b. HPLC conditions: Venusil MP C18 column (Agela Technologies, 5 μm, 10 × 250 mm), CH3CN (with 0.1% TFA)/H2O (with 0.1% TFA) = 70/30, 2 mL/min, UV 254 nm. HPLC co-elution profiles of 8a, 8b and 9a, 9b. HPLC conditions: Venusil MP C18 column (Agela Technologies, 5 μm, 10 mm × 250 mm), CH3CN/H2O = 90/10, 2 mL/min, UV 254 nm
In vitro stability study
Each radiotracer (1b, 2b, 3b, 8b and 9b) (100 μL, 100 μCi) was added in a centrifugal tube containing Kunming rat serum albumin (500 μL) respectively, then incubated at 37 °C for 3 h. The serum proteins were precipitated by adding 200 μL acetonitrile and centrifuged at 3500 rpm for 10 min. The supernatant was filtered with a 0.22 μm Millipore filter and the radiochemical purity was detected by HPLC (Fig. 3).

In vitro stability of 99mTc-labeled complexes in rat serum after incubating at 37 °C for 3 h. HPLC conditions: Venusil MP C18 column (Agela Technologies, 5 μm, 10 × 250 mm), CH3CN (with 0.1% TFA)/H2O (with 0.1% TFA) = 70/30, 2 mL/min, UV 254 nm. HPLC co-elution profiles of 8a, 8b and 9a, 9b. HPLC conditions: Venusil MP C18 column (Agela Technologies, 5 μm, 10 mm × 250 mm), CH3CN/H2O = 90/10, 2 mL/min, UV 254 nm
Determination of the partition coefficient
One milli litre of n-octanol, 0.9 mL of phosphate buffer (PBS; 0.1 M, pH 7.4) and 0.1 mL of (1b, 2b, 3b, 8b and 9b) was mixed in a test tube. After vortexing the centrifuge tube for 3 min at room temperature, the tube was centrifuged for 5 min at 3500 rpm. Two samples (0.1 mL) from the n-octanol and PBS were collected, weighted and counted. The partition coefficient was expressed as the logarithm of the ratio of the count per gram of n-octanol versus PBS. The measurement was repeated three times.
Biodistribution
When the animals are in the fast state, fatty acids are mainly metabolized in the heart. Based on this the mice were fasted for 12 h before experiment. Prepared radiotracers were dissolved in saline with 10% ethanol. About 100 μL (10 μCi, 3.7 × 102 Bq) of the five prepared radiotracers were injected into the mice by means of the tail vein. Different groups of animals (n = 5) were killed exactly at 1, 5, 15, 30, 60 min, and the interested organs and tissues (blood, heart, brain, lung, liver, spleen, kidney, muscle and bone) were collected, then weighed and counted.
Blookade study
8b was choose for blooking study. One group, the Re complex (at a dose of 100 nmol/0.1 mL) was co-injection with the 99mTc-radiotracer. Another group, the Re complex (at a dose of 100 nmol/0.1 mL) was injected 30 min prior to the 99mTc complex. The third group, 99mTc-radiotracter was injected with saline. The Kunming mice (female, 18–22 g) were prepared by pre-treating corresponding non-radioactivity Re complex which were killed at 1, 5, 15, 30 and 60 after the injected 99mTc-radiotracter. Heart was collected, weighed and counted.
Analysis of metabolites
Radiotracer 8b (100 µL/500 µCi, 18.5 MBq) was injected into normal mice (KM, female 30–35 g, n = 3) by intravenous administration, the mice were killed 30 min postinjection and heart tissue was collected. The hearts were placed in cold PBS (300 µL) and homogenized. The protein was precipitated using acetonitrile (300 µL). After centrifugation, the supernatant was analyzed using HPLC.
In vitro cytotoxicity assay
The standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in vitro was applied to assess cell toxicity using H9c2 cells. The H9c2 cells were plated onto each well and incubated at 37 °C in Dulbecco’s Modified Eagle Medium containing 10% FBS under an atmosphere of 4.5% CO2 for 24 h. The compound 8a of different concentrations (1 mL, 100, 80, 60, 40, 20, 10, 1 or 0.1 μmol/L) were added into different wells which incubating with H9c2 cells at 37 °C for 24 h. Then 10 μL of MTT (5 mg/mL) solution in PBS (pH 7.4) was added into each well and further incubated for 4 h. Cell lysates (100 μL/well) were added into each well and incubated overnight. Afterward, the absorbance was determined at 570 nm (for absorbance of MTT formazan) and 650 nm (for the reference wavelength). The results expressed as EC50 (concentration for 50% of maximal effect) were calculated using Graph Pad Prism software (San Diego, CA). Cell viability was calculated as a percentage of viable cells after being treated with compound 8a compared with the untreated cells.
Result and discussion
Synthesis and radiolabeling
The synthesis of the Re/99mTc are shown in Scheme3 and were characterized by Radio-HPLC using their well-characterized nonradioactive rhenium counterparts as references. The Radio-HPLC retention times of compound 1a.1b, 2a.2b, 3a.3b, 8a.8b and 9a.9b are summarized in Fig. 2 The retention times between 99mTc-labeled compounds and their Re counterparts exhibited small changes. The radiochemical purity was more than 95% after purification using HPLC. The overall radiochemical yield was 84.6 ± 5.2% (decay-corrected) for 1b, 2b, and 3b and was 16.8 ± 3.5% (decay-corrected) for 8b and 9b.
Stability study
The Tc-99m complexes prepared were incubated in rat serum for 3 h at 37 °C. The radio-HPLC analysis results (Fig. 3) showed that over 95% of radiotracers remained. They all had great stability which fit for developing as molecular imaging agents.
Partition coefficient (log D7.4)
The log D7.4values (Table 1) of 8b and 9b were found to be 1.75 ± 0.04 and 1.39 ± 0.01, respectively, falling within an appropriate logP value to enter the myocardium and both the five compounds were more soluble in liposoluble substances.
Blokade study
The results were showed in the Table 2. There was no effectively decrease in the heart uptake. The reason was 99mTc-labelled fatty acids were metabolic imaging agents, and no specific binding site existed. It was a big difference with receptor imaging agents.
Biodistribution
Table 3 showed the myocardial uptake, radioactivity profile in other organs and clearance of the radiotracers under evaluation. 99mTc-CpTT-16-oxo-HDA [15] was a classical 99mTc-labled fatty acid, the results of it was shown in Table 4 for comparison. The biodistribution studies in mice showed 8b had high myocardial uptake of 8.17% ID/g similar to the 99mTc-CpTT-16-oxo-HDA standard at 1 min, however with poor retention subsequently. For different animal species and experiment conditions, it is probably in appropriate for direct comparisons in % ID/g of radiotracers, the proper comparison between different species may be the heart to other non-target organ ratios (Fig. 4). 8b was eliminated rapidly in blood or lungs. The high initial uptake and retention in heart together with favorable heart/blood ratio suggest that pyrrole group of 8b is worthy of further study. For compound 2b, the heart-to-liver ratio (0.37 at 1 min postinjection) was observed to be better than the value obtained with 99mTc-CpTT-16-oxo-HDA at same interval of time. Radioactivity clearance from myocardium and other organs were found to clear with time. However, the ratios of heart-to-blood and heart-to-lung with those prepared complexes were significantly low in comparison with values obtained with 99mTc-CpTT-16-oxo-HDA. Compared to the FAs showed in the previously literature [20], 1b, 2b and 3b introduced furan ring replaced isothere thiopene ring, the heart uptake of 1b, 2b and 3b was improved, and 2b had prolonged myocardium uptake of 1.89% ID/g at 60 min postinjection. It came to conclusion that furan ring may benefit to be taken by heart. Compound 8b showed lower blood uptake and better clearance, compared to 1b, 2b or 3b. The reason may be that the molecular size of cyclopentadienyl tricarbonyl 99mTc core was smaller than N,N,N-donors incorporated monocationic [99mTc(CO)3]+ core. However, both of the two cores were worthy of study, for they had different characteristics.

Comparison of a Heart/blood ratio b Heart/lung ratio c Heart/liver ratio of 1b, 2b, 3b, 8b, 9b with99mTc-CpTT-16-oxo-HDA [15]
Metabolic analysis
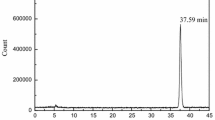
As previous literature reported, fatty acids are metabolized by β-oxidation losing two -CH2- groups [15], which lead short chain residues formed. The retention time on HPLC of short chain fatty acids are shorter comparing to long chain fatty acids using C-18 column (eluent: acetonitrile/water). Analysis results of radiotracers 8b in myocardium homogenate was showed in Fig. 5. The peak at retention time of 7.21 min was radiotracers 8b in analytical HPLC condition. It was observed that a peak at shorter retention time (5.25 min) may metabolite of 8b by β-oxidation. However, no more efforts were made to research the formation of the metabolite.

Metabolic analysis of myocardium homogenates collected at 30 min postinjection (Analytical column, Agela Venusil MP C18, 150 × 4.6 mm, 5 µm) using isocratic elution system (mobile phase A is water at a rate of 0 mL/min, while mobile phase B is acetonitrile at a rate of 0.7 mL/min) and UV (254 nm))
In vitro cytotoxicity assay
The standard MTT cells assay was employed using H9c2 cells to examine the cytotoxicity of compound 8a. The EC50 value was more than 100, which showed compound 8a was nearly nontoxic.
Conclusions
The pyrrol group is beneficial to decrease lung uptake and enhance blood clearance of Tc-99m-labeled fatty acid analogs and the introduction of amide may lead to obviously improved lung uptake. 8b was superior to other four 99mTc-FA in biodistribution studies, but 8b cannot be used as myocardial imaging agent for the poor heart-liver ratios which may provide a lad experience for technetium investigations aiming at activation of other fatty acid tracers. 1b, 2b and 3b are small-sized monocationic tricarbonyl technetium-labelled fatty acid analogs, the tissue distribution showed low radioactivity accumulation in liver with high heart-to-liver ratio. Cyclopentadienyl tricarbonyl 99mTc core is beneficial to reduce blood accumulation and high heart-to-blood ratio, compared to N,N,N-donors incorporated monocationic [99mTc(CO)3]+ core.
References
Corbett JR (1999) Fatty acids for myocardial imaging. J Semin Nucl Med 29:237–258
Van der Vusse GJ, Glatz JF, Stam HC, Reneman RS (1992) Fatty acid homeostasis in the normoxic and ischemic heart. Physiol Rev 72(4):881–940
Neely JR, Rovetto MJ, Oram JF (1972) Myocardial utilization of carbohydrate and lipids. Prog Cardiovasc Dis 15:289–329
Liedtke AJ (1981) Alterations of carbohydrate and lipid metabolism in the acutely ischemic heart. J Prog Cardiovasc Dis 23:321–336
Opie LH (1968) Metabolism of the heart in health and disease. J Am Heart 76:685–698
Neely JR, Whitmer KM, Mochizuki S (1976) Effects of mechanical activity and hormones on myocardial glucose and fatty acid utilization. J Circ Res 38:22–29
Van der Vusse GJ, Stam HC (1987) Lipid and carbohydrate metabolism in the ischaemic heart. J Basic Res Cardiol 82(suppl 1):149–153
Palaniswamy SS, Padma S (2011) Cardiac fatty acid metabolism and ischemic memory imaging with nuclear medicine techniques. Nucl Med Commun 32:672–677
Tamaki N, Morita K, Kuge Y, Tsukamoto E (2000) The role of fatty acids in cardiac imaging. J Nucl Med 41:1525–1534
Lerch R, Bergmann SR, Ambos HD, Welch MJ, Ter-Pogossian MM, Sobel BE (1982) Effect of flow independent reduction of metabolism on regional myocardial clearance of 11C-palmitate. Circulation 65:731–738
Wyns W, Schwaiger M, Huang SC, Buxton DB, Hansen H, Selin C, Keen R, Phelps ME (1989) Effects of inhibition of fatty acid oxidation on myocardial kinetics of 11C-palmitate. J Circ Res 65:1787–1797
Tamaki N, Kawamoto M, Yonekura Y, Fujibayashi Y, Takahashi N, Konishi J, Nohara R, Kambara H, Kawai C, Ikekubo K (1992) Regional metabolic abnormality in relation to perfusion and wall motion in patients with myocardial infarction: assessment with emission tomography using an iodinated branched fatty acid analog. J Nucl Med 33:659–667
Magata Y, Kawaguchi T, Ukon M, Yamamura N, Uehara T, Ogawa K, Arano Y, Temma T, Mukai T, Tadamura E, Saji H (2004) A Tc-99 m-labeled long chain fatty acid derivative for myocardial imaging. Bioconjugate Chem 15:389–393
Tomoya U, Tomoe U, Seiji H, Sayaka A, Kenichi O, Hiromichi A (2007) Technetium-99 m-labeled long chain fatty acid analogues metabolized by β-oxidation in the heart. J Med Chem 50:543–549
Lee BC, Kim DH, Lee I, Choe YS, Chi DY, Lee KH, Choi Y, Kim BT (2008) 16-Cyclopentadienyl tricarbonyl 99mTc 16-Oxo-hexadecanoic acid: synthesis and evaluation of fatty acid metabolism in mouse myocardium. J Med Chem 51:3630–3634
Zeng HH, Zhang HB (2014) Synthesis and biological evaluation of fatty acids conjugates bearing cyclopentadienyl-donors incorporated [99mTc/Re(CO)3]+ for myocardical imaging. Eur J Med Chem 72:10–17
Mathur A, Subramanian S, Mallia MB, Banerjee S, Samuel G, Sarma HD, Venkatesh M (2008) Synthesis and bio-evaluation of a new fatty acid derivative for myocardial imaging. Bioorgan Med Chem 16:7927–7931
Mathur A, Mallia MB, Sarma HD, Banerjee S, Venkatesh M (2011) Synthesis, radiolabeling and evaluation of a new positively charged 99mTc-labeled fatty acid derivative for myocardial imaging. J Label Compd Rad 54:150–156
Cazzola E, Benini E, Pasquali M, Mirtschink P, Walther M, Pietzsch HJ (2008) Labeling of fatty acid ligands with the strong electrophilic metal fragment [99mTc(N)(PNP)]2+(PNP = diphosphane ligand). Bioconjugate Chem 19:450–460
Xue QQ, Wang H, Liu JP (2016) Synthesis and biodistribution of novel dipicolylamine 99mTc-(CO)-labeled fatty acid derivatives for myocardial imaging. J Radioanal Nucl Chem 310:1181–1194
Toth G, Kover KE (1995) Simple, safe, large scale synthese of 5-arylmethyl-2,2-dimethyl-1,3-dioxane-4,6-dioanensd-3-aryl-propanoic acids. Synth Commun 25:3067–3074
Zhu X, Wang Y, Li HR (2011) Do all the protic ionic liquids exist as molecular aggregates in the gas phase. Phys Chem Chem Phys 13:17445–17448
Reddy VR, Mallireddigari MR, Cosenza SC et al (2008) Design, synthesis, and biological evaluation of (E)-Styrylbenzylsulfones as novel anticancer agents. J Med Chem 51:86–100
Liu JP, Xue QQ, Wang H, Wang H, Wang DW, Zhang HB (2016) Synthesis and bio-evaluation of Tc-99m-labeledfatty acid derivatives for myocardial metabolism imaging. Appl Organomet Chem 30:596–604
Lazarova N, James S, Babich J, Zubieta J (2004) A convenient synthesis, chemical characterization and reactivity of [Re(CO)3(H2O)3]Br: the crystal and molecular structure of [Re(CO)3(CH3CN)2Br]. Inorg Chem Commun 7:1023–1026
Spradau TW, Katzenellenbogen JA (1998) Preparation of cyclopentadienyl tricarbonylrhenium complexes using a double ligand-transfer reaction. Organometallics 17:2009–2017
Chen X, Cui MC (2012) Synthesis and biological evaluation of a novel 99mTc cyclopentadienyl tricarbonyl complex ([(Cp-R)99mTc(CO)3]) for sigma-2 receptor tumor imaging Bioorg. Med Chem Lett 22:6352–6357
Alberto R, Schibli R, Egli A (1998) A novel organometallic aqua complex of technetium for the labeling of biomolecules: synthesis of [99mTc(OH2)3(CO)3]+ from [99mTcO4]− in aqueous solution and its reaction with a bifunctional ligand. J Am Chem Soc 120:7987–7988
Acknowledgment
We gratefully acknowledged the Significant New Drugs Development (Grant No. 2014ZX09507007001), Natural Science Foundation of China (No. 21371026) and Science and Technology Support Program (No. 2014BAA03B03) for financial support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liu, J., Wang, S., Wang, H. et al. Synthesis and biological evaluation of fatty acid derivatives for myocardial imaging containing [99mTc(CO)3]+ . J Radioanal Nucl Chem 312, 543–555 (2017). https://doi.org/10.1007/s10967-017-5258-2
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-017-5258-2