
Abstract
Deficiency of human adenosine deaminase type 2 (DADA2) is a complex systemic autoinflammatory disorder characterized by vasculopathy, immune dysregulation, and hematologic abnormalities. The most notable neurological manifestations of DADA2 are strokes that can manifest with various neurological symptoms and are potentially fatal. However, neurological presentations can be diverse. We here present a review of the neurological manifestations of DADA2 to increase clinical awareness of DADA2 as the underlying diagnosis. We reviewed all published cases of DADA2 from 1 January 2014 until 19 July 2022 found via PubMed. A total of 129 articles describing the clinical features of DADA2 were included in the analysis. Six hundred twenty-eight patients diagnosed with DADA2 were included in the review. 50.3% of patients had at least signs of one reported neurological event, which was the initial or sole manifestation in 5.7% and 0.6%, respectively. 77.5% of patients with neurological manifestations had at least signs of one cerebrovascular accident, with lacunar strokes being the most common and 35.9% of them having multiple stroke episodes. There is a remarkable predilection for the brain stem and deep gray matter, with 37.3% and 41.6% of ischemic strokes, respectively. Other neurological involvement included neuropathies, focal neurological deficits, ophthalmological findings, convulsions, and headaches. In summary, neurological manifestations affect a significant proportion of patients with DADA2, and the phenotype is broad. Neurological manifestations can be the first and single manifestation of DADA2. Therefore, stroke, encephalitis, posterior reversible encephalopathy syndrome, mononeuropathy and polyneuropathy, and Behçet’s disease-like presentations should prompt the neurologist to exclude DADA2, especially but not only in childhood.
Similar content being viewed by others


Introduction
Deficiency of human adenosine deaminase type 2 (DADA2) was first described in 2014 by two independent groups as a condition characterized by fever, polyarteritis nodosa (PAN), livedo racemosa, early-onset stroke, liver disease, and mild immunodeficiency [1, 2]. Subsequently, the phenotype expanded to include additional hemato-immunological manifestations such as cytopenias, bone marrow failure, and hemophagocytosis [3,4,5]. The underlying cause of DADA2 is biallelic (homozygous or compound heterozygous) loss of function mutations in adenosine deaminase type 2 (ADA2), formerly known as Cat Eye Syndrome Chromosome Region 1 (CECR1) [1, 2].
DADA2 usually presents in childhood, with an average age of onset of 5 to 7 years; approximately 25% of patients have the onset of the symptoms before the age of 1 year and 77% by the age of 10 years [4, 6]. However, adult-onset has also been described [1, 2, 7,8,9]. In some cases, DADA2 can be misdiagnosed as PAN. Caorsi et al. described DADA2 in 15 (31%) out of 48 European children with early-onset PAN [10]. In adult patients with PAN, biallelic pathogenic ADA2 variants were detected in 4 out of 108 cases (3.4%) [11]. The incidence of DADA2 has been estimated to be around 1 in 222,000 individuals worldwide [12], with a mortality of 8% [4], mainly in childhood.
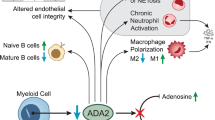
The pathophysiology of DADA2 remains incompletely understood [1, 2]. ADA2 is mainly but not exclusively expressed by myeloid cells, and its deficiency leads to impairment of endothelial integrity and the development of perivascular inflammation. Tumor necrosis factor (TNF) is a key cytokine involved in the pathological process but increased type I and II interferon (IFN) signaling have also been described [13, 14]. The monocyte differentiation in patients with DADA2 has been reported to be skewed, resulting in a decreased number of anti-inflammatory M2 macrophages and increased proinflammatory M1 macrophages [15]. ADA2 may play a role in the activation of neutrophils; thus, in ADA2-deficient patients, hyperactivation of neutrophils may cause endothelial damage [16]. Additionally, patients with DADA2 have been described to display an increased production of neutrophil extracellular traps (NETs) and subsequently increased TNF production [17].
Most likely, a dysregulation of the homeostasis of the vascular endothelium in the presence of proinflammatory macrophages leads to vessel stenosis, aneurysm formation, and perforation of vessels [6]. This process primarily affects small and medium vessels and manifests in various locations, including the skin, cranial and peripheral nerves, kidneys, intestine, and even testes [18]. Signs and symptoms of DADA2 range from non-specific signs, such as fever, and weight loss, to specific findings resembling PAN, such as nodular vasculitis, livedo racemosa, ulcers, abdominal pain, bowel perforation, and portal or arterial hypertension. Since the histopathological findings of vasculitis are indistinguishable between PAN and DADA2, the latter is sometimes classified as a subtype of “classic” PAN [1], representing a more aggressive variant.
Notably, neurological complications are common in DADA2 [19], with strokes being a prominent feature that can present with various neurological symptoms and carry a high risk of fatality [1, 2, 10, 20]. Other neurological findings or initial alternative diagnosis include transient ischemic attacks (TIAs) with insignificant magnetic resonance imaging (MRI) findings, posterior reversible encephalopathy syndrome (PRES)-like encephalopathies, sensorineural hearing loss, ophthalmologic abnormalities, and peripheral and cranial neuropathies [21, 22]. Neurological symptoms can precede other manifestations in the course of DADA2 or be a sole presenting feature [4, 6, 23, 24]. Given the significant neurological impact of DADA2 and the need for increased clinical awareness, this paper aims to provide a comprehensive review of the neurological manifestations associated with this condition. By summarizing the relevant cases reported in the current scientific literature, we aim to enhance understanding and facilitate early recognition and management of DADA2-related neurological complications.
Methods
We reviewed the available data on patients diagnosed with DADA2 published between 1 January 2014 and 19 July 2022. The search was conducted manually via PubMed using the following keywords: “ADA2 deficiency OR adenosine deaminase 2 deficiency OR DADA2.” After all articles describing the clinical features of DADA2 were included in the analysis. Articles that were not in English were not included in the study. Papers containing insufficient clinical descriptions of patients were excluded [3, 4, 6, 11, 12, 14, 17, 19, 25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87]. This was evaluated by full-text screening. In case of duplication of cases, only the most recent report or the report with more complete clinical description was included [88,89,90,91,92,93]. Two articles were not included because of unavailability of the full text [94, 95] (Fig. 1).

Flowchart of literature search
Results
A total of 628 patients diagnosed with DADA2 were included in the review to analyze the demographics, genotypes, and clinical characteristics.
Demographics
There were 221 (46.4% of patients with known sex) female patients, 255 (53.6%) male patients, and 152 patients whose sex was not specified.
Of 274 patients with reported ethnicity (43.6%), 137 were Caucasian (50%), 43 were South Asian (15.7%), 27 were East Asian (9.9%), 43 came from the Middle East (15.7%), 9 North African (3.3%), 9 Latin American (3.3%), 2 Jewish (0.7%), 2 African/Caucasian (0.7%), 1 African (0.4%), and 1 African American (0.4%).
The mean age of disease onset for 425 patients (67.7%) for whom it was reported was seven years. Thirty-three reported patients (7.8%) had an age of onset ranging between 18 and 59 [2, 9, 20, 96–105]. In 20 of these, the adult presentation was with stroke or another neurological manifestation.
Genetics
The notations of the patients’ mutations in the ADA2 gene were standardized according to the NM_001282225.2 transcript.
The pathogenic variants were reported for 495 patients (78.8%). Two hundred seventy-seven patients had a homozygous variant, 205 compound heterozygous, 12 patients had one heterozygous variant with an unknown second mutation, and one patient had both homozygous and heterozygous deletions. There were 82 missense mutations, 28 frameshift mutations, 9 splice site mutations, 7 nonsense mutations, 3 duplications, 18 deletions, 2 start loss mutations, 1 deletion-insertion mutation, and 1 unspecified intronic mutation. Figure 2 and Supplementary Table S1 present the genetic characteristics in detail.

Pathogenic variants of ADA2. Genomic sequence with exonic regions indicated by boxes and the corresponding protein with domains indicated below. Reported pathogenic variants are indicated above. SP, signal peptide; DD, dimerization domain; CD, catalytic domain; PRB D, putative receptor binding domain
Clinical Characteristics
Neurological Manifestations
Among the reviewed patients, 316 (50.3%) had at least one reported neurological event throughout the course of the disease. The mean age of onset of neurological manifestation was 7.2 years (range: 0–36 years). A total of 245 patients (77.5% of all neurological manifestations) had at least one cerebrovascular accident (CVA), including 115 ischemic, 61 hemorrhagic strokes, and five transient ischemic attacks; 88 patients (35.9%) had multiple stroke episodes. Thirty-four patients presented with neuropathy (19 patients with mononeuropathies and 15 patients with polyneuropathies). Other neurological involvements included focal neurological deficits (such as cranial nerve paralysis, decreased/increased tendon reflexes, upper motor neuron signs, numbness, extremity weakness, sensory loss in the lower extremity) (n=9), ophthalmological findings (such as strabismus, loss of vision) (n=2), convulsions (n=5), headache, including migraines (n=4), and aspecific findings like dizziness and amnesia (n=2). In addition, cerebral atrophy (n=4), white matter changes (n=2), radiographic findings of brain anomaly (n=1), and five patients with unspecified neurological findings were reported. The spectrum of neurological symptoms in patients with DADA2 is shown in Fig. 3.

The spectrum of neurological symptoms in patients with DADA2 (n=316). CVA, cerebrovascular accident; TIA, transient ischemic attack
For 18 patients (5.7% of patients with neurological involvement), a neurological event represented the initial presentation of DADA2 [2, 13, 21, 23, 24, E6–E14]; for two of them [2, 13], it was the sole manifestation of the disease, as reported in the literature. However, the majority of patients initially presented with a variety of hematological, immunodeficient, and vascular manifestations, such as recurrent fever, weight loss, recurrent upper airway infection, livedo racemosa or reticularis, skin and oral aphthous ulcers, Raynaud phenomenon, arterial hypertension, myocarditis, abdominal pain, diarrhea, hepatosplenomegaly, myalgia, arthritis, alopecia, hypogammaglobulinemia, cytopenia, testicular, mesenteric, pancreatic, splenic, and renal infarcts. It is worth noting that the existing literature and awareness of DADA2 largely stem from rheumatology and immunology studies, potentially resulting in skewed representation of neurological manifestations.
Twenty-two patients (3.5% of all the reviewed patients) had neurocognitive disorders [1, 8, 13, 21, 97, E9, E15–E21]. These included isolated cognitive impairment, isolated memory disturbance, autism spectrum disorder (ASD), attention deficit hyperactivity disorder (ADHD), isolated persistent irritability, and isolated difficulties in concentration. Most of the patients were reported to have only one type of neurocognitive disorders; only those diagnosed with ASD also had ADHD, and one patient with memory disturbance also had difficulties in concentration. The age of the disease onset of the patients with neurocognitive disorders ranges from 0 to 14 years, with the mean age of 4.3 years.
MRI findings of 146 patients were available (Table 1) [1, 2, 10, 13, 16, 20,21,22,23,24, 96, E1, E4–E7, E9–E11, E13–E15, E17–E19, E21–E51]. Other diagnostic reports for neurological involvement included computer tomography [2, E4, E52–E54], angiographic investigation (reveals arterial stenosis, aneurysms) [1, 2], nerve conduction study, and needle electromyography [E55] (both considered to serve as non-specific diagnostic tools of neuropathies in DADA2). According to the review of neuroimaging results, the most common CNS lesion during DADA2 is ischemic stroke, with most common localizations in the brain stem, thalamus, basal ganglia, internal capsule, and other deep cerebral structures, representing the pathological involvement of deep perforating arteries. Lacunar stroke cases comprise 62.7% of the available MRI findings, of which 90.2% were located in the deep gray matter or brain stem. Vascular wall enhancement of small perforating arteries has also been reported [E32], but may be under-represented because of the absence of vessel-wall-imaging sequences in studies. We note that the described imaging findings are based on MRI, which is the preferred imaging modality due to its ability to detect acute ischemic stroke using diffusion-weighted sequences and its suitability for comprehensive evaluation of intracranial manifestations in children due to the absence of ionizing radiation.
Discussion
In this survey of neurological manifestations of DADA2, we found that 50.3% of patients had at least one reported neurological event. The spectrum of neurological manifestations was broad: from ischemic and hemorrhagic strokes to peripheral neuropathy, neuritis, and neurocognitive disorders, such as ASD and learning difficulties.
The mean age of onset of neurological manifestation is 7.2 years, with the youngest patient being only 3 months old and the oldest being 36 years old at the time of neurological manifestations onset. Thus, the onset of neurological manifestations is in line with the onset of DADA2 manifestations in general (7 years), although adult-onset is possible. The late onset can lead to misdiagnosis; therefore, differential diagnosis of DADA2 is obligatory in every case of PAN [48].
Strokes account for 77.5% of all neurological events and, in some cases, can be the initial [2, 13, 21, 23, 24, E6–E14] or even sole [2, 13] manifestation of DADA2. Some neurological signs of CNS involvement can be underestimated due to their mild or transient character (i.e., TIA or “silent” infarcts) or due to the possibility of MRI-negative infarcts, particularly in the brainstem, which may be missed without the use of thin cuts or higher magnetic field strength. Thus, it is crucial to exclude DADA2 whenever a patient presents with an acute stroke at any age, especially in childhood. Besides other non-atherosclerotic arteriopathies, such as arterial dissection, focal cerebral arteriopathy, and Moyamoya vasculopathy, DADA2 should be in the differential diagnosis of arterial ischemic stroke in childhood [E56]. It is also noteworthy that in the past, many patients who would have been diagnosed with Sneddon syndrome are likely to be DADA2 patients.
Interestingly, the preference for the brain stem and deep gray matter structures on MRI is similar to the preferred locations in Behçet’s disease, another immune-mediated vasculitis, albeit that lesions in Behçet’s disease are often not infarcts. This parallel between the imaging pattern of a DADA2-mutation and Behçet’s disease has not been reported on to our knowledge. Yet, an overlap from a clinical perspective [E57] and genetic perspective [E58] has been described before.
Other neurological symptoms and syndromes, such as mononeuropathy and polyneuropathy, and PRES, are also sentinels of DADA2. Mononeuritis multiplex was diagnosed in 12 of the reviewed patients [2, 9, 20, E37, E59, E60]. Caorsi et al. reported that neuropathy of cranial and peripheral nerves was common in DADA2 and that these signs distinguished DADA2 from childhood PAN [10]. A recent paper by Ehlers L. et al. [E61] describes a case of encephalopathy and multifocal neurological deficits in a DADA2 patient previously diagnosed with acute disseminated encephalomyelitis (ADEM). Therefore, it is important to consider the possibility of DADA2 in such patients and maintain a reasonable level of suspicion.
Interestingly, intracranial calcifications, the hallmark of Aicardi-Guttieres syndrome (AGS), the archetypical interferonopathy, are typically absent in DADA2 patients. This is noteworthy considering the involvement of enhanced type I interferon signaling in DADA2 patients and the consideration of SAMHD1 and other AGS underlying genes in the differential diagnosis [16, E62]. The absence of intracranial calcification in DADA2 might suggest that the mechanisms underlying brain abnormalities in interferonopathies may not be applicable to those in DADA2. The other explanation would be that the level of type I IFN upregulation is much higher in typical AGS than in DADA2 [16]. In addition, the exact pathophysiological link between the increased type I IFN and the calcifications in AGS is incompletely understood. It is also important to note that not all interferonopathies are characterized by calcifications.
In addition, patients with DADA2 can have a broad spectrum of subtle neurocognitive disorders, which should be taken into consideration in patients with DADA2, and the evaluation of these conditions should be part of the routine management of DADA2. We hypothesize that the actual number of patients with neurocognitive disorders may be even higher than reported due to underdiagnosis. Our study shows that 22 DADA2 patients (3.5% of all the reviewed patients) had reported neurocognitive disorders. Contrasting the obtained data with the general population, there seems to be a higher prevalence of these disabilities in DADA2. According to a systemic review update by Zeidan J et al., 1% of children have ASD in the general population [E63, E64]. Sayal K et al. state that 5% of all children and adolescents are diagnosed with ADHD. Whether this is only a coincidence, or the discrepancy is due to a direct effect of ADA2 deficiency, or an indirect impact of the context of severe disease is unclear at this stage.
Although neuroimaging findings may show a wide variety, ranging from normal findings to PRES or ADEM-like presentations, the most common manifestation was ischemic and to lesser extent hemorrhagic stroke, presumably reflecting the underlying vasculitis. Especially, lacunar infarcts in the brain stem and deep gray matter were common, in line with previously reported data [1, E22, E32]. It is interesting that the preference for these specific localizations is similar to that of the neurological manifestation of Behçet’s disease, which also has a known clinical [E57] and genetic [E58] overlap with DADA2. Direct signs of underlying vasculitis have also been demonstrated with MRI with vessel wall imaging [E32].
Patients with minor or resolved neurologic symptoms are of particular concern, as half of them do have acute infarcts on MRI (foci of restricted diffusion on MRI) [E65]. They are, therefore, at risk of developing recurrent strokes and disabilities [E66]. The differential diagnosis of ischemic stroke, especially in children, presents a challenge for clinicians, with arteriopathies being the most common cause [80]. As neurological manifestations may lack specificity in DADA2, further investigation is needed to find specific biomarkers, particularly for assessing the risk of stroke, especially in the case of “mild” neurological signs. In the absence of a promising biomarker, the diagnosis of DADA2 requires a high index of suspicion and awareness of the wide phenotypic variability of the disease. We recommend formal exclusion of DADA2 as the underlying condition by both plasma ADA2 enzymatic function testing, as well as ADA2 sequencing [4]. In most patients with absent or severely reduced serum ADA2 enzyme activity, a bi-allelic loss-of-function mutation in the ADA2 gene is found [51].
Early diagnosis offers the opportunity for early initiation of targeted treatment with TNF-α inhibitors which can prevent long-term sequelae due to recurrent ischemic events. In the study of the efficacy of anti-TNF treatment for preventing strokes in patients with DADA2 conducted by Cooray et al., the authors reported the median rate of ischemic events in DADA2 patients to be 2.37 before anti-TNF treatment vs. 0.0 per 100 patient-months after treatment [E5]. Among the biological agents used, etanercept shows the most robust evidence [E31]. Anti-TNF treatment seems to be less effective in reversing the hematological manifestations though [78], E5, E27. Treatment-refractory patients can be cured by hematopoietic stem cell transplantation (HSCT) [81, 93, E67]. At this time, HSCT remains the only available curative option, although gene therapy is on the horizon. In two recent studies [E3, E68], a lentiviral vector-mediated hematopoietic stem and progenitor cells (HSPC) gene therapy has been described to restore ADA2 expression and enzymatic activity in patients’ CD34+ cells, normalize secretion of IL-6 and TNFα from macrophages, correcting the inflammatory macrophage phenotype. It is also important to note that aspirin (or other antiplatelet agents) is traditionally not recommended in DADA-2 because of a risk of brain hemorrhage [14, 78].
In conclusion, neurological manifestations affect a significant proportion of patients with DADA2, and the phenotype is broad. Neurological manifestations can be the first and single manifestation of DADA2. Therefore, stroke, encephalitis (including ADEM), PRES, mononeuropathy and polyneuropathy, and Behçet’s disease-like presentations should prompt the neurologist to exclude ADA2 deficiency, especially but not only in childhood. We are convinced that this review will aid in recognizing DADA2 early after the disease onset, which will allow for early treatment initiation and hopefully limit the detrimental outcomes often associated with DADA2.
Additionally, this study has several limitations. The study only included articles written in English, which may have excluded relevant data published in other languages, and therefore could have introduced a language bias into the study. The study only included published articles, which could have resulted in the exclusion of unpublished data or data published in less accessible sources. This could have introduced a publication bias into the study. The quality of the data included in the study could have been influenced by the quality of reporting in the articles reviewed. This could have introduced a reporting bias into the study. The study was a literature review and did not include any primary data collection, which could limit the depth and accuracy of the analysis.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Zhou Q, Yang D, Ombrello AK, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. 2014;370(10):911–20. https://doi.org/10.1056/NEJMoa1307361.
Navon Elkan P, Pierce SB, Segel R, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med. 2014;370(10):921–31. https://doi.org/10.1056/NEJMoa1307362.
Hashem H, Kelly SJ, Ganson NJ, Hershfield MS. Deficiency of adenosine deaminase 2 (DADA2), an inherited cause of polyarteritis nodosa and a mimic of other systemic rheumatologic disorders. Curr Rheumatol Rep. 2017;19(11):70. https://doi.org/10.1007/s11926-017-0699-8.
Meyts I, Aksentijevich I. Deficiency of adenosine deaminase 2 (DADA2): updates on the phenotype, genetics, pathogenesis, and treatment. J Clin Immunol. 2018;38(5):569–78. https://doi.org/10.1007/s10875-018-0525-8.
Signa S, Bertoni A, Penco F, et al. Adenosine deaminase 2 deficiency (DADA2): a crosstalk between innate and adaptive immunity. Front Immunol. 2022;13:935957. https://doi.org/10.3389/fimmu.2022.935957.
Lee PY. Vasculopathy, immunodeficiency, and bone marrow failure: the intriguing syndrome caused by deficiency of adenosine deaminase 2. Front Pediatr. 2018;6:282. https://doi.org/10.3389/fped.2018.00282.
Bourgeois G, Richard M, Danset M, Pérard L, Breton AL, Berthoux E. Deficiency of adenosine deaminase 2 diagnosed at 65 years of age. Lancet Lond Engl. 2021;397(10277):913. https://doi.org/10.1016/S0140-6736(20)32660-X.
Trotta L, Martelius T, Siitonen T, et al. ADA2 deficiency: clonal lymphoproliferation in a subset of patients. J Allergy Clin Immunol. 2018;141(4):1534–1537.e8. https://doi.org/10.1016/j.jaci.2018.01.012.
Sharma A, Naidu G, Sharma V, et al. Deficiency of adenosine deaminase 2 in adults and children: experience from India. Arthritis Rheumatol Hoboken NJ. 2021;73(2):276–85. https://doi.org/10.1002/art.41500.
Caorsi R, Penco F, Grossi A, et al. ADA2 deficiency (DADA2) as an unrecognised cause of early onset polyarteritis nodosa and stroke: a multicentre national study. Ann Rheum Dis. 2017;76(10):1648–56. https://doi.org/10.1136/annrheumdis-2016-210802.
Schnappauf O, Sampaio Moura N, Aksentijevich I, et al. Sequence-based screening of patients with idiopathic polyarteritis nodosa, granulomatosis with polyangiitis, and microscopic polyangiitis for deleterious genetic variants in ADA2. Arthritis Rheumatol. 2021;73(3):512–9. https://doi.org/10.1002/art.41549.
Jee H, Huang Z, Baxter S, et al. Comprehensive analysis of ADA2 genetic variants and estimation of carrier frequency driven by a function-based approach. J Allergy Clin Immunol. 2022;149(1):379–87. https://doi.org/10.1016/j.jaci.2021.04.034.
Nihira H, Izawa K, Ito M, et al. Detailed analysis of Japanese patients with adenosine deaminase 2 deficiency reveals characteristic elevation of type II interferon signature and STAT1 hyperactivation. J Allergy Clin Immunol. 2021;148(2):550–62. https://doi.org/10.1016/j.jaci.2021.01.018.
Barron KS, Aksentijevich I, Deuitch NT, et al. The spectrum of the deficiency of adenosine deaminase 2: an observational analysis of a 60 patient cohort. Front Immunol. 2021;12:811473. https://doi.org/10.3389/fimmu.2021.811473.
Zavialov AV, Gracia E, Glaichenhaus N, Franco R, Zavialov AV, Lauvau G. Human adenosine deaminase 2 induces differentiation of monocytes into macrophages and stimulates proliferation of T helper cells and macrophages. J Leukoc Biol. 2010;88(2):279–90. https://doi.org/10.1189/jlb.1109764.
Belot A, Wassmer E, Twilt M, et al. Mutations in CECR1 associated with a neutrophil signature in peripheral blood. Pediatr Rheumatol. 2014;12(1):44. https://doi.org/10.1186/1546-0096-12-44.
Carmona-Rivera C, Khaznadar SS, Shwin KW, et al. Deficiency of adenosine deaminase 2 triggers adenosine-mediated NETosis and TNF production in patients with DADA2. Blood. 2019;134(4):395–406. https://doi.org/10.1182/blood.2018892752.
Clarke K, Campbell C, Omoyinmi E, et al. Testicular ischemia in deficiency of adenosine deaminase 2 (DADA2). Pediatr Rheumatol. 2019;17(1):39. https://doi.org/10.1186/s12969-019-0334-5.
Kasap Cuceoglu M, Sener S, Batu ED, et al. Systematic review of childhood-onset polyarteritis nodosa and DADA2. Semin Arthritis Rheum. 2021;51(3):559–64. https://doi.org/10.1016/j.semarthrit.2021.04.009.
Özen S, Batu ED, Taşkıran EZ, et al. A monogenic disease with a variety of phenotypes: deficiency of adenosine deaminase 2. J Rheumatol. 2020;47(1):117–25. https://doi.org/10.3899/jrheum.181384.
Westendorp WF, Nederkoorn PJ, Aksentijevich I, Hak AE, Lichtenbelt KD, Braun KPJ. Unexplained early-onset lacunar stroke and inflammatory skin lesions: consider ADA2 deficiency. Neurology. 2015;84(20):2092–3. https://doi.org/10.1212/WNL.0000000000001581.
Varghese A, Thomas J, Maramattom B. ‘More than just skin in the game’. DADA2 autoinflammatory syndrome and stroke in the young. Ann Indian Acad Neurol. 2021;24(3):410 https://doi.org/10.4103/aian.AIAN_716_20.
Elbracht M, Mull M, Wagner N, et al. Stroke as initial manifestation of adenosine deaminase 2 deficiency. Neuropediatrics. 2016;48(02):111–4. https://doi.org/10.1055/s-0036-1597611.
Gburek-Augustat J, Platzer K, Schumann I, et al. Case report: Deficiency of adenosine deaminase 2 (DADA2) as a cause of brainstem stroke in a 3-year-old girl and the importance of early fast-track genetic diagnostics to influence therapy. Neuropediatrics. 2022;53(06):432–5. https://doi.org/10.1055/a-1896-5817.
Fayand A, Chasset F, Boutboul D, et al. DADA2 diagnosed in adulthood versus childhood: a comparative study on 306 patients including a systematic literature review and 12 French cases. Semin Arthritis Rheum. 2021;51(6):1170–9. https://doi.org/10.1016/j.semarthrit.2021.09.001.
Kastner DL, Zhou Q, Aksentijevich I. Mutant ADA2 in vasculopathies. N Engl J Med. 2014;371(5):480–1. https://doi.org/10.1056/NEJMc1405506.
Segel R, King MC, Levy-Lahad E. Mutant ADA2 in vasculopathies. N Engl J Med. 2014;371(5):481. https://doi.org/10.1056/NEJMc1405506.
Stoffels M, Kastner DL. Old dogs, new tricks: monogenic autoinflammatory disease unleashed. Annu Rev Genomics Hum Genet. 2016;17(1):245–72. https://doi.org/10.1146/annurev-genom-090413-025334.
Karadag O, Erden A, Bilginer Y, et al. A retrospective study comparing the phenotype and outcomes of patients with polyarteritis nodosa between UK and Turkish cohorts. Rheumatol Int. 2018;38(10):1833–40. https://doi.org/10.1007/s00296-018-4122-1.
Sönmez HE, Batu ED, Taşkıran EZ, Alikaşifoğlu M, Bilginer Y, Özen S. Genetic testing for DADA2: how can we avoid missing patients? Eur J Hum Genet. 2018;26(11):1563–5. https://doi.org/10.1038/s41431-018-0240-1.
Santo GC, Baldeiras I, Guerreiro R, et al. Adenosine deaminase two and immunoglobulin M accurately differentiate adult Sneddon’s syndrome of unknown cause. Cerebrovasc Dis. 2018;46(5-6):257–64. https://doi.org/10.1159/000495794.
Lee PY. NETing the mechanism of inflammation in DADA2. Blood. 2019;134(4):338–9. https://doi.org/10.1182/blood.2019001251.
Zervou M, Goulielmos G, Matalliotakis M, Matalliotaki C, Spandidos D, Eliopoulos E. Role of adenosine deaminase 2 gene variants in pediatric deficiency of adenosine deaminase 2: a structural biological approach. Mol Med Rep. Published online December. 2019;5 https://doi.org/10.3892/mmr.2019.10862.
Baytaroğlu A, Kadayifçilar S, Ağin A, et al. Choroidal vascularity index as a biomarker of systemic inflammation in childhood Polyarteritis Nodosa and adenosine deaminase-2 deficiency. Pediatr Rheumatol. 2020;18(1):29. https://doi.org/10.1186/s12969-020-0417-3.
Schnappauf O, Ombrello AK, Kastner DL. Deficiency of adenosine deaminase 2: is it an elephant after all? J Allergy Clin Immunol. 2020;145(6):1560–1. https://doi.org/10.1016/j.jaci.2020.04.023.
Mesa-Nuñez C, Mortellaro A. T cells and monocytes: a dangerous liaison in adenosine deaminase 2 deficiency. J Leukoc Biol. 2022;111(2):297–9. https://doi.org/10.1002/JLB.3CE1021-561R.
Vinchi F. Towards a cure for adenosine deaminase 2 deficiency through genetic correction of macrophage polarization. HemaSphere. 2021;5(11):e653. https://doi.org/10.1097/HS9.0000000000000653.
Wu Z, Gao S, Watanabe N, et al. Single-cell profiling of T lymphocytes in deficiency of adenosine deaminase 2. J Leukoc Biol. 2022;111(2):301–12. https://doi.org/10.1002/JLB.5A0621-314R.
Cafaro A, Pigliasco F, Barco S, et al. A novel LC–MS/MS-based method for the diagnosis of ADA2 deficiency from dried plasma spot. Molecules. 2021;26(18):5707. https://doi.org/10.3390/molecules26185707.
Yamashita M, Morio T. Another exciting data—HCT successfully cured patients with DADA2: a commentary on “Hematopoietic cell transplantation cures adenosine deaminase 2 deficiency: report on 30 patients” by Hashem H et al. J Clin Immunol. 2021;41(7):1443–5. https://doi.org/10.1007/s10875-021-01121-4.
Ito M, Nihira H, Izawa K, Yasumi T, Nishikomori R, Iwaki-Egawa S. Enzyme activity in dried blood spot as a diagnostic tool for adenosine deaminase 2 deficiency. Anal Biochem. 2021;628:114292. https://doi.org/10.1016/j.ab.2021.114292.
Kaya Akca U, Sag E, Unal S, Kasap Cuceoglu M, Bilginer Y, Ozen S. The role of vascular inflammation markers in deficiency of adenosine deaminase 2. Semin Arthritis Rheum. 2021;51(4):839–44. https://doi.org/10.1016/j.semarthrit.2021.04.013.
Ehlers L, Meyts I. What a difference ADA2 makes: insights into the pathophysiology of ADA2 deficiency from single-cell RNA sequencing of monocytes. J Leukoc Biol. 2021;110(3):405–7. https://doi.org/10.1002/JLB.5CE0421-186R.
Zervou M, Goulielmos G, Matalliotakis M, Matalliotaki C, Spandidos D, Eliopoulos E. [Corrigendum] Role of adenosine deaminase 2 gene variants in pediatric deficiency of adenosine deaminase 2: a structural biological approach. Mol Med Rep. 2021;24(1):489. https://doi.org/10.3892/mmr.2021.12128.
Naidu G, Kopp CR, Sharma V, et al. Validation of the provisional seven-item criteria for the diagnosis of polyarteritis nodosa. Rheumatol Int. 2021;41(9):1651–5. https://doi.org/10.1007/s00296-021-04867-7.
Schwartz DM, Kitakule MM, Dizon BL, et al. Systematic evaluation of nine monogenic autoinflammatory diseases reveals common and disease-specific correlations with allergy-associated features. Ann Rheum Dis. 2021;80(6):788–95. https://doi.org/10.1136/annrheumdis-2020-219137.
Schena F, Penco F, Volpi S, et al. Dysregulation in B-cell responses and T follicular helper cell function in ADA2 deficiency patients. Eur J Immunol. 2021;51(1):206–19. https://doi.org/10.1002/eji.202048549.
Caorsi R, Penco F, Schena F, Gattorno M. Monogenic polyarteritis: the lesson of ADA2 deficiency. Pediatr Rheumatol. 2016;14(1):51. https://doi.org/10.1186/s12969-016-0111-7.
Berkun Y, Segel R, Navon-Elkan P. Adenosine deaminase 2 deficiency: more than monogenic vasculitis. Isr Med Assoc J IMAJ. 2017;19(7):435–7.
Jain A, Misra DP, Sharma A, Wakhlu A, Agarwal V, Negi VS. Vasculitis and vasculitis-like manifestations in monogenic autoinflammatory syndromes. Rheumatol Int. 2018;38(1):13–24. https://doi.org/10.1007/s00296-017-3839-6.
Human A, Pagnoux C. Diagnosis and management of ADA2 deficient polyarteritis nodosa. Int J Rheum Dis. 2019;22:69–77. https://doi.org/10.1111/1756-185X.13283.
Mackay MT, Steinlin M. Recent developments and new frontiers in childhood arterial ischemic stroke. Int J Stroke. 2019;14(1):32–43. https://doi.org/10.1177/1747493018790064.
Alghamdi M. Autoinflammatory disease-associated vasculitis/vasculopathy. Curr Rheumatol Rep. 2018;20(12):87. https://doi.org/10.1007/s11926-018-0788-3.
Özen S, Sönmez HE, Demir S. Pediatric forms of vasculitis. Best Pract Res Clin Rheumatol. 2018;32(1):137–47. https://doi.org/10.1016/j.berh.2018.09.007.
Ozen S. What’s new in autoinflammation? Pediatr Nephrol. 2019;34(12):2449–56. https://doi.org/10.1007/s00467-018-4155-4.
Moens L, Hershfield M, Arts K, Aksentijevich I, Meyts I. Human adenosine deaminase 2 deficiency: a multi-faceted inborn error of immunity. Immunol Rev. 2019;287(1):62–72. https://doi.org/10.1111/imr.12722.
Leavis H, Zwerina J, Manger B, Fritsch-Stork RDE. Novel developments in primary immunodeficiencies (PID)—a rheumatological perspective. Curr Rheumatol Rep. 2019;21(10):55. https://doi.org/10.1007/s11926-019-0854-5.
Sahin S, Adrovic A, Kasapcopur O. A monogenic autoinflammatory disease with fatal vasculitis: deficiency of adenosine deaminase 2. Curr Opin Rheumatol. 2020;32(1):3–14. https://doi.org/10.1097/BOR.0000000000000669.
Sönmez HE, Karaaslan C, de Jesus AA, et al. A clinical score to guide in decision making for monogenic type I IFNopathies. Pediatr Res. 2020;87(4):745–52. https://doi.org/10.1038/s41390-019-0614-2.
Navallas M, Inarejos Clemente EJ, Iglesias E, Rebollo-Polo M, Zaki FM, Navarro OM. Autoinflammatory diseases in childhood, part 1: monogenic syndromes. Pediatr Radiol. 2020;50(3):415–30. https://doi.org/10.1007/s00247-019-04536-9.
Pain CE. Juvenile-onset Behçet’s syndrome and mimics. Clin Immunol. 2020;214:108381. https://doi.org/10.1016/j.clim.2020.108381.
Huang Z, Li T, Nigrovic PA, Lee PY. Polyarteritis nodosa and deficiency of adenosine deaminase 2 – shared genealogy, generations apart. Clin Immunol. 2020;215:108411. https://doi.org/10.1016/j.clim.2020.108411.
Sag E, Demir S, Ozen S. Clusters in pediatric rheumatic diseases. Curr Rheumatol Rep. 2020;22(7):28. https://doi.org/10.1007/s11926-020-00908-5.
Ozen S, Sag E. Childhood vasculitis. Rheumatology. 2020;59(Supplement_3):iii95–iii100. https://doi.org/10.1093/rheumatology/kez599.
Brogan P, Eleftheriou D. Vasculitis update: pathogenesis and biomarkers. Pediatr Nephrol. 2018;33(2):187–98. https://doi.org/10.1007/s00467-017-3597-4.
Lachmann HJ. Periodic fever syndromes. Best Pract Res Clin Rheumatol. 2017;31(4):596–609. https://doi.org/10.1016/j.berh.2017.12.001.
Demir S, Sag E, Dedeoglu F, Ozen S. Vasculitis in systemic autoinflammatory diseases. Front Pediatr. 2018;6:377. https://doi.org/10.3389/fped.2018.00377.
Aksentijevich I, Sampaio Moura N, Barron K. Adenosine deaminase 2 deficiency. In: Adam MP, Everman DB, Mirzaa GM, et al., editors. GeneReviews®. Seattle: University of Washington; 1993. http://www.ncbi.nlm.nih.gov/books/NBK544951/. Accessed 11 Jan 2023.
Karadag O, Jayne DJ. Polyarteritis nodosa revisited: a review of historical approaches, subphenotypes and a research agenda. Clin Exp Rheumatol. 2018;36 Suppl 111(2):135-142.
Flinn AM, Gennery AR. Adenosine deaminase deficiency: a review. Orphanet J Rare Dis. 2018;13(1):65. https://doi.org/10.1186/s13023-018-0807-5.
Kendall JL, Springer JM. The many faces of a monogenic autoinflammatory disease: adenosine deaminase 2 deficiency. Curr Rheumatol Rep. 2020;22(10):64. https://doi.org/10.1007/s11926-020-00944-1.
Springer JM, Byram K. Polyarteritis nodosa: an evolving primary systemic vasculitis. Postgrad Med. Published online June. 2022;22:1–8. https://doi.org/10.1080/00325481.2022.2088940.
Pilania RK, Banday AZ, Sharma S, et al. Deficiency of human adenosine deaminase type 2 – a diagnostic conundrum for the hematologist. Front Immunol. 2022;13:869570. https://doi.org/10.3389/fimmu.2022.869570.
Lee PY, Aksentijevich I, Zhou Q. Mechanisms of vascular inflammation in deficiency of adenosine deaminase 2 (DADA2). Semin Immunopathol. 2022;44(3):269–80. https://doi.org/10.1007/s00281-022-00918-8.
Bilginer Y, Ozen S. Polyarteritis nodosa. Curr Opin Pediatr. 2022;34(2):229–33. https://doi.org/10.1097/MOP.0000000000001106.
Mastrangelo M, Ricciardi G, Giordo L, Michele MD, Toni D, Leuzzi V. Stroke and stroke-like episodes in inborn errors of metabolism: pathophysiological and clinical implications. Mol Genet Metab. 2022;135(1):3–14. https://doi.org/10.1016/j.ymgme.2021.12.014.
Maccora I, Marrani E, Mastrolia MV, et al. Ocular involvement in monogenic autoinflammatory disease. Autoimmun Rev. 2021;20(11):102944. https://doi.org/10.1016/j.autrev.2021.102944.
Zhang B, Xu N, Chen J, et al. Treatment and outcome in deficiency of adenosine deaminase 2: a literature review. J Investig Allergy Clin Immunol. 2022;32(1):13–22. https://doi.org/10.18176/jiaci.0748.
López-Nevado M, González-Granado LI, Ruiz-García R, et al. Primary immune regulatory disorders with an autoimmune lymphoproliferative syndrome-like phenotype: immunologic evaluation, early diagnosis and management. Front Immunol. 2021;12:671755. https://doi.org/10.3389/fimmu.2021.671755.
Pizzatto R, Resende LL, Lobo CFT, et al. Arteriopathy in pediatric stroke: an underestimated clinical entity. Arq Neuropsiquiatr. 2021;79(4):321–33. https://doi.org/10.1590/0004-282x-anp-2020-0105.
Pinto B, Deo P, Sharma S, Syal A, Sharma A. Expanding spectrum of DADA2: a review of phenotypes, genetics, pathogenesis and treatment. Clin Rheumatol. 2021;40(10):3883–96. https://doi.org/10.1007/s10067-021-05711-w.
Conticini E, Sota J, Falsetti P, et al. Biologic drugs in the treatment of polyarteritis nodosa and deficit of adenosine deaminase 2: a narrative review. Autoimmun Rev. 2021;20(4):102784. https://doi.org/10.1016/j.autrev.2021.102784.
Lin YC, Kalot MA, Husainat NM, et al. Polyarteritis nodosa: a systematic review of test accuracy and benefits and harms of common treatments. ACR Open Rheumatol. 2021;3(2):91–100. https://doi.org/10.1002/acr2.11189.
Kaljas Y, Liu C, Skaldin M, et al. Human adenosine deaminases ADA1 and ADA2 bind to different subsets of immune cells. Cell Mol Life Sci. 2017;74(3):555–70. https://doi.org/10.1007/s00018-016-2357-0.
Collison J. Stem cell transplantation for DADA2. Nat Rev Rheumatol. 2017;13(12):694. https://doi.org/10.1038/nrrheum.2017.175.
Putaala J. Ischemic stroke in young adults. Contin Lifelong Learn Neurol. 2020;26(2):386–414. https://doi.org/10.1212/CON.0000000000000833.
Demir S, Sönmez HE, Özen S. Vasculitis: decade in review. Curr Rheumatol Rev. 2018;15(1):14–22. https://doi.org/10.2174/1573397114666180726093731.
Bucciol G, Delafontaine S, Segers H, et al. Hematopoietic stem cell transplantation in ADA2 deficiency: early restoration of ADA2 enzyme activity and disease relapse upon drop of donor chimerism. J Clin Immunol. 2017;37(8):746–50. https://doi.org/10.1007/s10875-017-0449-8.
Debatin KM. HSCT cures ADA2 deficiency. Blood. 2017;130(24):2582–3. https://doi.org/10.1182/blood-2017-10-811125.
Sönmez HE, Armağan B, Ayan G, et al. Polyarteritis nodosa: lessons from 25 years of experience. Clin Exp Rheumatol. 2019;37(Suppl 117(2)):52–6.
Ulirsch JC, Verboon JM, Kazerounian S, et al. The genetic landscape of Diamond-Blackfan anemia. Am J Hum Genet. 2018;103(6):930–47. https://doi.org/10.1016/j.ajhg.2018.10.027.
Suri D, Rawat A, Jindal AK, et al. Spectrum of systemic auto-inflammatory diseases in India: a multi-centric experience. Front Immunol. 2021;12:630691. https://doi.org/10.3389/fimmu.2021.630691.
Hashem H, Vatsayan A, Gupta A, Nagle K, Hershfield M, Dalal J. Successful reduced intensity hematopoietic cell transplant in a patient with deficiency of adenosine deaminase 2. Bone Marrow Transplant. 2017;52(11):1575–6. https://doi.org/10.1038/bmt.2017.173.
Signa S, Dell’Orso G, Gattorno M, Faraci M. Hematopoietic stem cell transplantation in systemic autoinflammatory diseases - the first one hundred transplanted patients. Expert Rev Clin Immunol. 2022;18(7):667–89. https://doi.org/10.1080/1744666X.2022.2078704.
Mandviwala MM, Verbsky JW, Khammar AJ, Walsh RD. Internuclear ophthalmoplegia in a child with deficiency of adenosine deaminase 2. J Neuroophthalmol. 2021;Publish Ahead of Print; https://doi.org/10.1097/WNO.0000000000001354.
Xu Y, Shan Y, Hu Y, et al. Case report: An adult patient with deficiency of adenosine deaminase 2 resembled unilateral frosted branch angiitis. Front Med. 2021;8:642454. https://doi.org/10.3389/fmed.2021.642454.
Schepp J, Proietti M, Frede N, et al. Screening of 181 patients with antibody deficiency for deficiency of adenosine deaminase 2 sheds new light on the disease in adulthood. Arthritis Rheumatol Hoboken NJ. 2017;69(8):1689–700. https://doi.org/10.1002/art.40147.
Michniacki TF, Hannibal M, Ross CW, et al. Hematologic manifestations of deficiency of adenosine deaminase 2 (DADA2) and response to tumor necrosis factor inhibition in DADA2-associated bone marrow failure. J Clin Immunol. 2018;38(2):166–73. https://doi.org/10.1007/s10875-018-0480-4.
Rama M, Duflos C, Melki I, et al. A decision tree for the genetic diagnosis of deficiency of adenosine deaminase 2 (DADA2): a French reference centres experience. Eur J Hum Genet EJHG. 2018;26(7):960-971. :https://doi.org/10.1038/s41431-018-0130-6
Chasset F, Fayand A, Moguelet P, et al. Clinical and pathological dermatological features of deficiency of adenosine deaminase 2: a multicenter, retrospective, observational study. J Am Acad Dermatol. 2020;83(6):1794–8. https://doi.org/10.1016/j.jaad.2020.03.110.
Funding
Isabelle Meyts is a Senior Clinical Investigator at the Research Foundation – Flanders and is supported by the KU Leuven C1 Grant (C16/18/007), the FWO Grant (G0E8420N), the Jeffrey Modell Foundation, and the European Research Council (ERC) (the European Union’s Horizon 2020 research and innovation program – grant agreement No. 948959).
 Rik Lories is a Senior Clinical Investigator at the Research Foundation – Flanders (FWO 18B2122N), KU Leuven Grants (C14/20/117, DOAC14/19/092, IDN/20/019), FWO Grants (G099922N and G0B2120N), the European Innovative Medicines-2 grant Hippocrates and the Flemish Institute of Biotechnology (VIB) Grand Challenges project Spartacus. Rik Schrijvers is a Senior Clinical Investigator at the Research Foundation – Flanders (FWO 1805518N) and supported by FWO Grant (G054022). Lisa Ehlers is supported by an FWO PhD fellowship (11E0123N).
Rik Lories is a Senior Clinical Investigator at the Research Foundation – Flanders (FWO 18B2122N), KU Leuven Grants (C14/20/117, DOAC14/19/092, IDN/20/019), FWO Grants (G099922N and G0B2120N), the European Innovative Medicines-2 grant Hippocrates and the Flemish Institute of Biotechnology (VIB) Grand Challenges project Spartacus. Rik Schrijvers is a Senior Clinical Investigator at the Research Foundation – Flanders (FWO 1805518N) and supported by FWO Grant (G054022). Lisa Ehlers is supported by an FWO PhD fellowship (11E0123N).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Isabelle Meyts was responsible for conceptualization and supervision. Material preparation, data collection, and analysis were performed by Mariia Dzhus, Benjamin Verhaaren, Marco Baggio, and Leen Moens. The first draft of the manuscript was written by Mariia Dzhus and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publishing
Not applicable.
Conflict of Interest
Isabelle Meyts has received research funding at UZ Leuven from CSL Behring, unrelated to this study. She also participates as an advisory board member for Boehringer-Ingelheim, through LRD KU Leuven. Leuven Research and Development, the technology transfer office of KU Leuven, has received consultancy and speaker’s fees, or research grants on behalf of Rik Lories from Abbvie, Amgen (formerly Celgene), Biosplice Therapeutics (formerly Samumed), Eli-Lilly, Galapagos, Janssen, MSD, Novartis, Pfizer, Sandoz, UCB, and Viatris. This study is supported by the ERN-RITA.
Additional information
Note: additional references can be found in supplementary materials
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dzhus, M., Ehlers, L., Wouters, M. et al. A Narrative Review of the Neurological Manifestations of Human Adenosine Deaminase 2 Deficiency. J Clin Immunol 43, 1916–1926 (2023). https://doi.org/10.1007/s10875-023-01555-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-023-01555-y