
Abstract
The precipitation of cementite (M3C) from as-quenched martensite during tempering at 500 and 700 °C was investigated in a Fe–1C–1Cr (wt%) alloy. Tempering for a short duration at 700 °C results in a Cr/Fe ratio in the core region of M3C precipitates which is equal to the bulk alloy composition, while a shell on the surface of the precipitates exhibits a higher Cr concentration. With a prolonged tempering up to 5 h, the shell concentration gradually increases toward the equilibrium value, but the core region has not yet reached the equilibrium value. After tempering for 5 s at 500 °C, there is no Cr enrichment found at the M3C–matrix interface, while a transition to partitioning of Cr is found during the first 5 min of tempering at 500 °C. These experimental results indicate that M3C grows without significant partitioning of substitutional elements at both temperatures initially, i.e., growth is carbon diffusion controlled. This stage is, however, very short, and soon after 5 s at 700 °C and 5 min at 500 °C, Cr diffusion becomes important. Calculations using the diffusion simulation software DICTRA and precipitation simulation software TC-PRISMA were performed. The diffusion simulations using the local equilibrium interface condition show excellent agreement with experiments concerning Cr enrichment of the particles, but the size evolution is overestimated. On the other hand, the precipitation simulations underestimate the size evolution. It is suggested that a major improvement in the precipitation model could be achieved by implementing a modified nucleation model that considers nucleation far from the equilibrium composition.
Similar content being viewed by others


Introduction
Since as-quenched ferrous martensitic materials are generally quite hard and brittle, they are usually subjected to a tempering treatment to improve the toughness before being used in applications. During tempering, precipitation of various carbides or nitrides accompanied by microstructural recovery or recrystallization controls the development of the properties such as strength, toughness, and creep resistance in both plain carbon and alloy steels [1,2,3,4,5,6,7]. Cementite (M3C) is one of the important types of precipitates due to its strong effect on mechanical properties by depleting carbon from the matrix and potential precipitation hardening [4,5,6,7,8], and moreover, precipitation of M3C during tempering is strongly related to the evolution of other types of carbides in alloy steels [8,9,10,11,12], and it has therefore attracted plenty of attention [1,2,3,4,5, 8,9,10,11].
The precipitation process can be divided into three stages, i.e., nucleation, growth and coarsening, but these stages are often overlapping and are frequently studied as concurrent processes [2, 3]. In the early stages of precipitation, the focus of this report, the nucleation rate and diffusion are generally rate controlling [5, 6]. In binary Fe–C steels, the growth of M3C is controlled by the coupled diffusion of C and Fe [1, 3, 13,14,15,16,17], while in Fe–C–X ternary steels, the added substitutional element generally retards the growth of M3C significantly [10, 18,19,20,21,22]. When modeling the growth of M3C, it is considered important to impose the right local condition at the interface between matrix and precipitates [3, 10]. One of the following three assumptions is generally used: (1) partitioning local equilibrium (PLE) where local equilibrium is maintained at the interface, (2) para-equilibrium (PE) [6, 7] where only carbon is at equilibrium at the interface and the precipitate inherits the substitutional solute content from the matrix and (3) non-partitioning local equilibrium (NPLE) [5] where the growing phase also inherits the substitutional solute content but grows under local equilibrium at the interface for both carbon and substitutional solutes. Growth under NPLE means that a sharp spike of substitutional solutes is being pushed in front of the moving interface. Applied to M3C precipitation from ferrite, it has previously been presented that at low temperatures (160–575 °C), for example, in Fe–C–Mn, Fe–C–Si, Fe–C–Si–Mn and Fe–C–Mn–Cr–Mo–Si–Ni alloys, initial growth of M3C occurs under PE conditions [3, 9, 22,23,24,25,26,27]. However, it has also been reported that M3C formation in a Fe–0.6C–2Si alloy at 450 °C is controlled by neither PE nor NPLE evidenced by rejection of Si from the initial nucleus of M3C [3]. The nucleation of M3C is generally modeled using classical nucleation theory (CNT). A central assumption of CNT is the “capillarity approximation,” which means that the nucleating phase has the same properties, e.g., molecular density, chemical potential and surface tension, as it would have in its bulk form. Furthermore, the interface separating the nucleus and the parent phases is approximated as a sharp, infinitely thin barrier, and the nucleus shape is spherical (circular in 2D) [6, 9, 11, 28, 29]. It is still today challenging to understand how these assumptions affect the precipitation modeling and how it could be improved [28,29,30].
The above-mentioned reports on low-temperature tempering of M3C suggest that nucleation and growth depends on the temperature and alloying, leading to different compositions of the initial nucleus and the early growth is controlled by PE, while later stages of growth are influenced by the diffusion of substitutional elements to and within the M3C. With increasing temperature, the controlling mechanism of growth has been proposed to change from PE to NPLE [10, 22, 30], and in the case of pearlitic and upper-bainitic phase transformations this transition from PE to NPLE has been reported [10, 22, 31]. In the case of M3C precipitation, a systematic experimental and modeling work is lacking to clarify the interface condition during early stages of precipitation of M3C at high tempering temperatures. The existing studies have either focused on modeling [3] or are limited to measurements of the average chemical composition of precipitates [32].
The purpose of the present work is to experimentally investigate the early stages of precipitation of M3C, including the chemical composition at the interface between M3C and the matrix, in a Fe–1C–1Cr model alloy tempered at 500 and 700 °C. Simulations using the software programs DICTRA and TC-PRISMA [33,34,35] were performed to complement the experiments.
Materials and methods
Alloy preparation and heat treatment
The studied alloy was prepared by vacuum induction melting followed by hot-rolling into 10-mm-thick plates. The chemical composition of the alloy is presented in Table 1. Specimens (10 × 10 × 1 mm) were austenitized at 1100 °C for 10 min followed by quenching in brine. Martensite with a negligible fraction of retained austenite was obtained [36]. Thereafter, the as-quenched samples were tempered at (1) 700 °C and (2) 500 °C for 5 s, 5 min, 30 min and 5 h, followed by quenching in brine. In order to eliminate the heating time, a Sn-Bi metal bath was employed for the short time heat treatments, i.e., 5 s, 5 min and 30 min. The 5-h-tempered samples were encapsulated in evacuated quartz tubes, and the tempering was performed in a muffle furnace.
Microstructure characterization
Samples for microstructure characterization were ground and mechanically polished, finishing with polishing using 0.05 μm Al2O3 slurry. The general microstructure evolution was characterized using backscattered imaging in a JEOL JSM-7800F scanning electron microscope (SEM), operating at 15 kV. The identification and quantification of the precipitates were conducted using carbon extraction replicas [36] and a JEOL JEM-2100F transmission electron microscope (TEM) operating at 200 kV, mostly under scanning transmission electron microscope (STEM) mode. For each tempered sample, approximately 1000 precipitates were imaged and their size was evaluated by assuming spherical particles and evaluating the sphere radius from 2D projected with an equivalent areas in STEM images [37]. Energy-dispersive X-ray spectroscopy (EDS) analysis was performed in TEM for over 50 precipitates for each tempering condition and the chemical composition of the precipitates was evaluated.
To study the elemental distribution at the interface between matrix and M3C and within the carbide, lamellae TEM samples were prepared by focused ion beam (FIB) sectioning from the 5-s-, 5-min-, 30-min- and 5-h-tempered samples at 700 °C and the 5-h-tempered sample at 500 °C. These conditions were the only conditions where the size of the precipitates enabled a quantitative measurement of the composition of the precipitates. During FIB preparation, it was assured that the thickness of the FIB thin foil was thinner than the thickness of the precipitate of interest for further TEM analysis. For the other conditions, only electro-polished thin foil for qualitative analysis of the composition was prepared since the size of the precipitates was sub-50 nm, preventing quantitative analysis. A FEI Nova NanoLab 600 dual-beam system, equipped with an Omniprobe micromanipulator in situ lift-out probe, at an acceleration voltage of 30 kV with varying beam currents of the Ga ion-source, was used to perform the FIB cross-sectioning; the final cleaning was performed with a 5 kV ion beam. The details for the preparation of electro-polished thin foil for 500 °C-tempered samples (5 s, 5 min, 30 min) can be found in [37]. TEM-EDS analysis and imaging were performed in a FEI Titan Themis Cs probe-corrected microscope operating at 200 kV. Thickness measurements were performed on these FIB and electro-polished thin foil using electron energy loss spectroscopy (EELS) for both precipitates and matrix. For 700 °C-tempered samples, a fairly uniform thickness of 75 nm in 5-s- and 5 min-tempered samples, 100 nm in 30-min-tempered sample and 125-nm in 5-h-tempered sample were achieved. For 500 °C-tempered samples, the thickness is 50 nm for all thin foils, while a thickness of 75 nm in the 5 h FIB thin foil was obtained. The difference in thickness between the middle of the measured precipitate and the surrounding matrix is less than 15% for all samples, and thus, EDS absorption correction can be applied to these measurements using the Cliff-Lorimer thin-film approach [38].
To determine the volume fraction of precipitates, the matrix phase was selectively dissolved in 10%AA (10v/v % acetylacetone–1 w/v % tetramethylammonium chloride–methanol) electrolyte, using a current of 40–60 mA and a charge of 1000–1200 coulombs with platinum electrodes [39, 40]. After electrolytic extraction, the solution was filtered using a polycarbonate filter (PCF) with an open-pore size of 50 nm. Then, careful weighing with the accuracy of ± 10−5 g was performed to find the amount of the extracted precipitates after they were completely dried. The weight of the metal sample was measured before and after extraction to determine the total dissolved weight. Standard errors were calculated from four sets of measurements of the weight of PCF and the sample. The residue carbides were analyzed by X-ray diffraction (XRD) using a Bruker D8 Discover diffractometer with a LynxEye 1D detector. The XRD measurements were performed in Bragg–Brentano geometry using Cu-Kα radiation. The diffraction patterns were recorded from 30 to 90° 2θ with angular steps of 0.01° and a dwell time of 6 s per step. The size of the precipitates was evaluated by the modified Williamson–Hall (WH) method from the peak broadening of XRD peaks. The analysis procedure was adopted from Refs. [41, 42].
Kinetic models
The thermodynamic calculations, diffusion simulations and precipitation simulations were performed using the Thermo-Calc software package including the diffusion (DICTRA) and precipitation (TC-PRISMA) modules with databases TCFE8 and MOBFE3 [33,34,35]. In DICTRA, PLE at the moving interface is assumed [33], but it is also possible to perform simulations under PE condition.
The growth stage of M3C was simulated using DICTRA with a single cell system, composed of a martensitic matrix (BCC-α) and one M3C nucleus. The geometry of M3C was assumed to be spherical. The radius of the critical nucleus r* can be estimated assuming an interfacial energy of M3C/BCC-α at the initial stage. Different interfacial energies (γ) between 0.1 and 0.3 J m−2 have been adopted in the literature [43, 44], and this leads to an assumed supercritical nucleus size of M3C of R1 = 1 nm in the present work at both 500 °C and 700 °C.
The radius of the matrix R3 can be calculated by the relationship between the calculated equilibrium volume fraction (\( V_{f}^{\text{cem}} \)) of M3C and the radius of M3C (R2) obtained from experimental work given as:
The composition of the small cementite particle was set to the same Cr/Fe ratio as for the matrix phase. The interfacial energy for the growth stage was 0.18 J/m2, calculated using the Becker’s model as implemented in TC-PRISMA. For simplicity, only the elements Fe, Cr and C were considered in the simulations.
The software TC-PRISMA was used to predict the full M3C precipitation event, i.e., nucleation, growth and coarsening. The models implemented in TC-PRISMA are described in [34, 35, 45]. The so-called simplified growth model was used, which means that the tie-line across the bulk composition is used throughout the simulation. This is different from the situation in DICTRA where an iterative procedure is used to calculate a new operating tie-line for each time step. There is an advanced growth model implemented in TC-PRISMA, which accounts for the change in operating tie-line, but unfortunately simulations using this model were not possible. The nucleation sites were set as dislocations due to the martensitic microstructure of the as-quenched samples, and an initial value of 1.2 × 1015 m−2 was used as taken from literature for similar microstructures [37, 41]. The input parameters including nucleation site type, dislocation density, and interfacial energy were varied within reasonable ranges to study the influence on the simulation results. The interfacial energy of M3C/BCC-α was varied from 0.1 to 1 J m−2, and the dislocation density was varied from 1014 to 1016 m−2 [41].
Results and discussion
Microstructure evolution
Figure 1 shows the microstructure of the studied alloy after tempering for different durations at 700 °C. After 5 s of tempering, the microstructure consists of plate-shaped martensite with many planar defects and thus there is no significant change from the as-quenched one. It is also clearly shown that the precipitates have been nucleated at boundaries, planar defects as well as inside plates, see Fig. 1a, a1, a2. This indicates that the nucleation of precipitates is very fast. Between 5 and 30 min of tempering, the precipitates grow and the number density decreases, see Fig. 1c, d. After 5 h of tempering, large M3C are mostly located at various boundaries. The microstructure evolution at 500 °C is qualitatively similar to that observed at 700 °C, but the rate of evolution is slower.

Microstructure and precipitates evolution during tempering at 700 °C. a 5 s; a1 and a2 are enlarged parts in a; b 5 min; c 30 min; d 5 h
Figure 2 shows SEM images and XRD patterns of extracted precipitates from different conditions. The results of the XRD analysis show only the presence of M3C-type carbides throughout the whole tempering at both temperatures. This is consistent with the equilibrium phases calculated using Thermo-Calc and is also confirmed by selected area electron diffraction (SAED) patterns in the TEM (not shown). Comparison of the XRD patterns from the different tempered conditions shows clear peak broadening in samples tempered at 500 °C for 5 s and 5 h and for samples tempered at 700 °C for 5 s, see Fig. 2a. This peak broadening is related to the fine size of M3C at these stages. At 700 °C, a clear peaks shift toward lower angles is also found after tempering for 30 min and longer durations as compared to the 5 s sample (see the 210 and 103 peak shifts in Fig. 2b). Similar peak shifts but with smaller degree are also found at 500 °C.

SEM images a, b and c XRD patterns of extracted precipitates at different tempering durations at 700 and 500 °C, d enlargement of the 2θ region from 40° to 50° shown in c for a clear view of the peak shift
TEM-EDS line profile analysis across M3C-matrix interface in the samples tempered for 5 s, 5 min, 30 min and 5 h at 700 °C are presented in Fig. 3. After 5 s, the Cr/Fe ratio in the center of M3C is equal to that in the bulk alloy, while at the M3C–matrix interface an enrichment of Cr is found. This enrichment results in a zone depleted of Cr near the M3C–matrix interface as shown in Fig. 3a. It is worth noting that the Cr/Fe ratio in the matrix phase is not at the alloy composition level some tens of nanometers away from the precipitates. This may be related to the high density of particles leading to an influence from nearby particles. The Cr concentration at the M3C–matrix interface has not reached the predicted equilibrium level as estimated from Thermo-Calc calculations. EDS results from several precipitates of varying radii reveal a similar trend. This aforementioned information is also confirmed by EDS results from the carbon replica analyses (not shown) where there is no influence on the quantitative analysis from the matrix.

TEM-EDS line-scan profile across the M3C–matrix interface in the 5 s (a) 5 min (b) 30 min (c) and 5 h (d) tempered samples at 700 °C. Inset gives the bright field image with the EDS scan direction marked with white line across the M3C carbide. Blue dotted vertical line represents the interface between M3C and matrix, black dotted line represents the equilibrium value 0.0825 of Cr/Fe ratio in M3C, calculated by Thermo-Calc software, and black dashed line represents the Cr/Fe ratio in the bulk composition 0.0116. SD: Standard deviation of the current EDS measurements
After tempering for 5 min and 30 min at 700 °C, the Cr/Fe ratio both at the interface and in the middle of the precipitates has increased, see in Fig. 3b, c, but they are both still lower than the equilibrium value. After tempering for 5 h at 700 °C, the Cr/Fe ratio at the interface has reached the equilibrium value, while in the middle of the precipitates it is still lower than the equilibrium value. For some of the precipitates investigated, the difference in Cr/Fe between the middle and interface of the precipitate were rather small, which indicates that these precipitates nucleated earlier and have already reached close to equilibrium composition throughout. These observations are consistent with the XRD peak shifts in Fig. 2.
It should be noted that there are uncertainties in the chemical composition analysis using EDS, since the spatial resolution is a function of the interaction volume of the electron beam within the sample. A diffuse interface with a width of 5 to 15 nm was found in all the measurements shown in Figs. 3 and 5. This is due to reasons such as curved or inclined interface underneath the surface of the thin foil. To minimize this effect, it was made sure that the selected interfaces were at an edge-on condition with the horizontal plane. This was performed by ensuring a sharp contrast of the interface in the bright field and dark field images. Nonetheless, the diffuse interfaces in Figs. 3 and 5 indicate the achieved resolution of about 5 to 15 nm. Considering that, e.g., the NPLE condition can be atomistically sharp, we therefore refrain from drawing conclusions that rely on very high spatial resolution of the chemical analysis.
The PSD (particle size distribution) of M3C at 700 °C is presented in Fig. 4, and it can be seen that the nucleation stage is already completed after 5 s, since there are no new smaller particles forming after that tempering time. This is supported by the measured number density of particles, which shows a slight decrease after 5 s. Thus, from 5 s and onward the precipitation process should be growth-dominated before coarsening dominates after about 5 h [45].

Particle size distribution (PSD) of M3C at 700 °C for a 5 s, b 5 min, c 30 min. The total number density of M3C during tempering are: 1.164 × 1021 m−3 at 5 s, 4.66 × 1020 m−3 at 5 min, 6.97 × 1019 m−3 at 30 min
The evolution of the Cr distribution across the matrix–M3C interface in samples tempered at 500 °C is presented in Fig. 5. After tempering for 5 s (see Fig. 5a), no enrichment of Cr is found, indicating non-partitioning of Cr during growth of M3C at this stage. A slight enrichment of Cr is observed in the sample tempered for 5 min (see Fig. 5b), which is also verified by the mapping of the elemental distributions using TEM-EDS (not shown). After tempering for 30 min (see Fig. 5c), an apparent enrichment of Cr was found at the edge of precipitates, but the Cr/Fe ratio in the middle of the precipitate is still close to that of the bulk composition. This pile up of Cr is presumably caused by the diffusivity difference of Cr in BCC-matrix phase and cementite, i.e., 4.6 × 10−18 m−2 s and 7.8 × 10−21 m−2 s, respectively [33]. After tempering for 5 h, the enrichment of Cr has increased but has still not reached the calculated equilibrium values neither at the edge nor in the middle of the precipitate, see Fig. 5d.

EDS line-scan profile across the M3C–matrix interface in the 5 s (a) 5 min (b) 30 min (c) and 5 h (d) tempered samples at 500 °C. Inset gives the TEM image with the EDS scan direction marked with line across the M3C carbide. Black dotted line represents the equilibrium value 0.09016 of Cr/Fe ratio in M3C, calculated by Thermo-Calc software, and black dashed line represents the Cr/Fe ratio in the bulk composition 0.0116. SD Standard deviation of the current EDS measurements
Simulations of M3C precipitation during tempering
TC-PRISMA
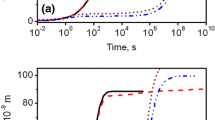
The simulated evolution of the mean radius and the volume fraction of M3C precipitates as a function of time from TC-PRISMA are shown and compared with experiments in Fig. 6. The mean size of the M3C is significantly underestimated in the initial stage of tempering, while the growth rate at the late stage is overestimated when the interfacial energy and dislocation density are varied within reasonable ranges. Similar results were obtained for the M3C evolution at 500 °C.

a Mean radius; b volume fraction; c mean radius at different nucleation sites and different dislocation density; d nucleation rate, at 700 °C
DICTRA
As already mentioned in Sect. 3, the TC-PRISMA simulations were performed assuming PLE conditions at the matrix–M3C interface with a static tie-line, and this limits the possibilities to account for the influence of different interface conditions on the growth rate. Therefore, DICTRA simulations, where the interface conditions could be varied between PE and PLE together with an operating tie-line changing with time, were performed.
At 700 °C, the radius of the matrix, R3, was calculated to 306 nm using Eq. 1 with the measured mean radius of M3C (R2) after 5 h tempering equal to 157 nm. The corresponding radii at 500 °C were R3 = 18 nm for R2 = 10 nm (mean radius after 5 h). These settings were done in order to ensure that a sufficiently large simulation cell was used and that the overall composition was preserved for the respective tempering temperature. The starting radius for M3C at both temperatures was set to 1 nm.
The DICTRA simulation at 700 °C, assuming PLE at the phase interface, can be seen in Fig. 7. The evolution of mean radius is little affected by including the Gibbs–Thomson effect (with interfacial energy set to 0.18 J/m2), see Fig. 7a, and the simulations are far from the experimental measurements. The growth of cementite is obviously controlled by the carbon diffusion in the very early stages, since chromium diffuses so much slower, leading to a spike of Cr being pushed in front of the growing phase, see Fig. 7b. This seems to be in qualitative accordance with the experimental findings (see Fig. 3), although the height and width of the Cr spike is different. In Fig. 7c, the variation in Cr/Fe ratio in cementite at different times has been summed. The corresponding summed mean values from the EDS measurements have also been included for comparison. It is interesting to see the excellent agreement between simulation and experiments in Fig. 7c, which gives some support to the thermodynamic and kinetic databases used but also to the assumed PLE conditions at the interface.

DICTRA simulation results for M3C growth during tempering of 700 °C: a mean radius, changes of Cr b at the M3C–matrix interface, average amount of Cr/Fe in the M3C at different times (c). The color bar on the top of the plot area represents the initial settings for the modeling and the interface positions. The inset in Fig. 7 a, b is the enlarged part of the selected area
In an attempt to account for the experimental results, the DICTRA simulations at 500 °C were performed with the PE model in DICTRA for the first 300 s, since the enrichment of Cr then seems to be very low or negligible (see Fig. 5a, c), and then changed to the default PLE conditions at the interface. The purpose of this modeling setup is to try and reproduce the experimental observations on the precipitation. It can be seen in Fig. 8a that the growth rate of the precipitates under the default PLE condition almost overlap with the PE condition, i.e., the change of interfacial condition is less important for the size evolution but important for the Cr enrichment. The results for the precipitate size evolution at 500 °C are in similar agreement with the experiments as the simulations at 700 °C, see Figs. 7 and 8. However, the concentration of Cr in the cementite is underestimated and it indicates that the diffusivity of Cr is significantly higher than the MOB3 database values, possibly due to a large effect of diffusion aided by defects, which is not a pronounced effect at 700 °C.

DICTRA simulation results for M3C during tempering at 500 °C: a mean radius, b changes of Cr (b) at the M3C–matrix interface, average amount of Cr/Fe in the M3C with tempering time (c). PE Para-equilibrium, PLE partitioning local equilibrium. The color bar on the top of the plot area represents the initial settings for the modeling and the interface positions. The inset in Fig. 8b is the enlarged part of selected area
Discussion on improved modeling of M3C precipitation
It is clear from the present work that neither the particle size nor the volume fraction evolution of M3C precipitates is captured well by the applied models. Therefore, we try to describe the reasons for these discrepancies between modeling and experiments here.
Classical nucleation theory (CNT) as implemented in TC-PRISMA is limited in that the composition of the critical nucleus is derived from the parallel tangent construction, which gives the highest driving force for nucleation. This composition is higher than the equilibrium composition of the precipitate, see, e.g., Hillert [46]. In fact, Hillert [46] showed that the more supersaturated the matrix phase is the higher the composition of the nucleus needs to be according to the parallel tangent construction. However, as seen in the experimental results the Cr composition of the cementite upon nucleation is far from the equilibrium composition, and at 500 °C after 5 s it is even close to the matrix phase composition, i.e., which can be called PE. But, the Cr composition at PE is lower than the equilibrium composition of the cementite and not higher as given by the parallel tangent construction. Hence, it seems the nucleation model needs to be modified to be able to account for PE conditions. However, by this modification of the critical nucleus composition, the nucleation event should become easier and therefore even more rapid than the current modeling results. On the other hand, the lowered driving force should lead to fewer but coarser precipitates, which seems promising in order to bring the simulation results closer to the experimental results. Growth should also be affected by a changed nucleation model, and it is predicted that the growth phase would slow down due to the additional diffusion needed when the precipitates nucleate with compositions far from the equilibrium composition. Another feature that would improve the physical nature of the model is to implement a treatment of the diffusion inside the cementite, as suggested by Zamberger et al. [11]. It should also be noted that the model assumption of spherical precipitates disagree with the experimentally observed elongated precipitates initially. The elongated particles should have faster kinetic evolution due to a larger Gibbs–Thomson effect and a shape effect [47, 48]. The elongated precipitates should be considered in further work.
As indicated in the above discussion, we suggest that a key feature to improve precipitation modeling would be to focus on the nucleation model. The DICTRA simulations do not account for the nucleation, but it is worthwhile to discuss the DICTRA predictions for the pure growth stage and the growth-dominated stage. According to the experimental information nucleation is already over after 5 s (Fig. 4) and pure coarsening initiates at about 5 h. This means that the growth-dominated stage from the experiments is several hours, whereas the DICTRA simulations indicate that the growth stage is about 0.01 s for both temperatures. Possibly this is due to that we only consider one single particle in the DICTRA simulations and that the multi-particle situation in reality leads to, e.g., overlapping diffusion fields (soft impingement) influencing the growth rate. Despite this, at 700 °C the DICTRA simulations are able to capture the evolution of Cr in the cementite (Fig. 7c), which supports the assumption of PLE at the moving interface. It should be pointed out that in DICTRA, due to the relatively low diffusivity of Cr in the matrix and M3C, the conditions at the interface during very early stages resemble NPLE. Hence, the change of operating tie-line seems to be an important factor for the correct growth of M3C at 700 °C. However, at 500 °C the change from PE to PLE was handled by manually switching model assumptions at the interface, which together with more uncertain diffusivities lead to a less good agreement with experiments (Fig. 8c).
Conclusions
1. The size and volume fraction of M3C increase extremely fast in the first 5 s of tempering at both 500 °C and 700 °C. The growth of M3C is controlled by carbon diffusion in the early stages.
2. At 700 °C, the Cr/Fe ratio in the middle of the M3C precipitates, after the first 5 s, is equal to the bulk Cr/Fe ratio, while the Cr enrichment at the interface zone indicates that redistribution of Cr has started. At 500 °C, no Cr enrichment was observed across the whole precipitate after 5 min of tempering, and an apparent Cr enrichment at the interface and in the middle of M3C precipitates was observed after 30 min.
3. At 500 °C, the conditions at the matrix–M3C interfaces evolve from close PE to PLE.
4. The size evolution of M3C is underestimated by the simulations in TC-PRISMA and overestimated by in DICTRA. It is suggested that a modified nucleation model in TC-PRISMA would improve the physical nature of the model significantly and improve predictive power.
References
Olson GB, Owen WS (1992) Martensite. ASM International, Materials Park, pp 263–269
Prat O, García J, Rojas D, Sanhueza JP, Camurri C (2014) Study of nucleation, growth and coarsening of precipitates in a novel 9% Cr heat resistant steel: experimental and modelling. Mater Chem Phys 143:754–764
Miyamoto G, Oh JC, Hono K, Furuhara T, Maki T (2007) Effect of partitioning of Mn and Si on the growth kinetics of cementite in tempered Fe–0.6 mass% C martensite. Acta Mater 55:5027–5038
Lv ZQ, Sun SH, Wang ZH, Qv MG, Jiang P, Fu WT (2008) Effect of alloying elements addition on coarsening behavior of pearlitic cementite particles after severe cold rolling and annealing. Mater Sci Eng A 489:107–112
Coates DE (1973) Diffusional growth limitation and hardenability. Metall Trans 4:2313–2325
M. Hillert: “Paraequilibrium,” Internal Report, Swedish Institute for Metal Research, Stockholm, 1953, appeared in Thermodynamics and Phase Transformations, The Selected Works of Mats Hillert, J. Ågren, Y. Bréchet, C. Hutchinson, J. Philibert, and G. Purdy, (Eds), EPD Science, Les Ulis, Cedex, France, 2006, pp 9–24
Hultgren A (1947) Isothermal transformation of austenite. Trans ASM 39:915–1005
Gladman T (1998) Principles of microstructural stability in creep resistant alloys, in book: Microstructural stability of creep resistant alloys for high temperature plant applications. IOM Communications, London, pp 1950–1968
Song WW, Choi PP, Inden G, Prahl U, Raabe D, Bleck W (2014) On the spheroidized carbide dissolution and elemental partitioning in high carbon bearing steel 100Cr6. Metall Mater Trans A 45:595–606
Zamberger S, Whitmore L, Krisam S, Wojcik T, Kozeschnik E (2015) Experimental and computational study of cementite precipitation in tempered martensite. Model Simul Mater Sci Eng 23(5):055012
Martin JW, Doherty RD (1976) Stability of microstructure in metallic systems: Cambridge solid state science series. Cambridge University Press, Cambridge, pp 219–366
Vedula KM, Heckel RW (1970) Spheroidization of binary Fe–C alloys over a range of temperatures. Metall Trans 1:9–18
Lindsley BA, Marder AR (1998) The morphology and coarsening kinetics of spheroidized Fe–C binary alloys. Acta Mater 46:341–351
Lifshitz IM, Slyosov VV (1961) The kinetics of precipitation from supersaturated solid solutions. J Phys Chem Solids 19:35–50
Oriani RA (1964) Ostwald ripening of precipitates in solid matrices. Acta Metall 12:1399–1409
Li CY, Blakely JM, Feingold AH (1966) Mass transport analysis for Ostwald ripening and related phenomena. Acta Metall 14:1397–1402
Mukherjee T, Stumpf WE, Sellars CM, Tegart WJM (1969) Kinetics of coarsening of carbides in chromium steels at 700 °C. J Iron Steel Inst 207:621–631
Schneider A, Inden G (2005) Simulation of the kinetics of precipitation reactions in ferritic steels. Acta Mater 53:519–531
Inden G (2008) Kinetics of phase transformations in multi-component systems. In: Ghetta V, Gorse D, Mazière D, Pontikis V (eds) Materials issues for generation IV systems. NATO Science for peace and security series. Springer, Heidelberg, pp 113–140
Björklund S, Donaghey LF, Hillert M (1972) The effect of alloying elements on the rate of Ostwald ripening of cementite in steel. Acta Metall 20:867–874
Barrow ATW, Kang JH, Rivera-Díaz-del-Castillo PEJ (2012) The ϵ → η→θ transition in 100Cr6 and its effect on mechanical properties. Acta Mater 60:2805–2815
Thomson RC, Miller MK (1998) Carbide precipitation in martensite during the early stages of tempering Cr- and Mo- containing low alloys steels. Acta Mater 46:2203–2213
Thomson RC, Miller MK (1996) An atom probe study of cementite precipitation in autotempered martensite in an Fe–Mn–C alloy. Appl Surf Sci 94/95:313–319
Babu SS, Hono K, Sakurai T (1993) APFIM studies on martensite tempering of Fe–C–Si–Mn low alloy steel. Appl Surf Sci 67:321–327
Zhu C, Xiong XY, Cerezo A, Hardwicke R, Krauss G, Smith GDW (2007) Three–dimensional atom probe characterization of alloy element partitioning in cementite during tempering of alloy steel. Ultramicroscopy 107:808–812
Clarke AJ, Miller MK, Field RD, Coughlin DR, Gibbs PJ, Clarke KD, Alexander DJ, Powers KA, Papin PA, Krauss G (2014) Atomic and nanoscale chemical and structural changes in quenched and tempered 4340 steel. Acta Mater 77:17–27
Vehkamäki H (2006) Classical nucleation theory in multicomponent systems. Springer, Berlin, pp 85–118
Hickey J, L’Heureux I (2013) Classical nucleation theory with a radius-dependent surface tension: a two-dimensional lattice-gas automata model. Phys Rev E 87:022406
Kelton KF, Frenkel D (2016) Preface: special topic on nucleation: new concepts and discoveries. J Chem Phys 145:211501
Song WW, Appen J, Choic P, Dronskowski R, Raabe D, Bleck W (2013) Atomic-scale investigation of ε and θ precipitates in bainite in 100Cr6 bearing steel by atom probe tomography and ab initio calculations. Acta Mater 61:7582–7590
Klein S, Mujica Roncery L, Walter M, Weber S, Theisen W (2017) Diffusion processes during cementite precipitation and their impact on electrical and thermal conductivity of a heat-treatable steel. J Mater Sci 52:375–390. https://doi.org/10.1007/s10853-016-0338-1
The Diffusion module (DICTRA) User Guide 2017a. Accessed 30 Aug 2017
TC-PRISMA. http://www.thermocalc.com/products-services/software/precipitation-module(TC-PRISMA). Accessed 2018
Precipitation Module (TC-PRISMA) User Guide. http://www.thermocalc.com/media/40970/precipitation-module-TC-PRISMAuser-guide.pdf. Accessed 30 Aug 2017
Hou ZY, Hedström P, Xu YB, Wu D, Odqvist J (2014) Microstructure of martensite in Fe–C–Cr and its implications for modelling of carbide precipitation during tempering. ISIJ Int 54:2649–2656
Hou ZY, Hedström P, Xu YB, Wu D, Odqvist J (2016) Quantitative modeling and experimental verification of carbide precipitation in a martensitic Fe–0.16 wt%C–4.0 wt% Cr alloy. Calphad 53:39–48
Chen YQ, Prasath Babu R, Slater TJA, Bai MW, Mitchell R, Ciuca O, Preuss M, Haigh SJ (2016) An nvestigation of diffusion-mediated cyclic coarsening and reversal coarsening in an advanced Ni-based superalloy. Acta Mater 110:295–305
Asadabad MA, Kheirandish S, Novinrooz AJ (2010) Microstructural and mechanical behavior of 4.5Cr-2 W-0.25 V- 0.1C steel. Mater Sci Eng A 527:1612–1616
Rivas AL, Vidal E, Matlock DK, Speer JG (2008) Electrochemical extraction of microalloy carbides in Nb-steel. Rev Metal 44:447–456
Takebayashi S, Kunieda T, Yoshinaga N, Ushioda K, Ogata S (2010) Comparison of the dislocation density in martensitic steels evaluated by some X-ray diffraction methods. ISIJ Int 50:875–882
Hou ZY, Babu P, Hedström P, Odqvist J (2018) Microstructure evolution during tempering of martensitic Fe–C–Cr alloys at 700 °C. J Mater Sci 53:6939–6950. https://doi.org/10.1007/s10853-018-2036-7
Sonderegger B, Kozeschnik E (2009) Generalized nearest neighbor–broken bond analysis of randomly oriented coherent interfaces in multi-component fcc and bcc structures. Metall Mater Trans A 40:499–510
Sonderegger B, Kozeschnik E, Leitner H, Clemens H, Svoboda J, Fischer FD, Staron P (2010) Kinetics of precipitation in a complex hot-work tool steel. Steel Res Int 81:64–73
Kampmann R, Wagner R (1984) Kinetics of precipitation in metastable binary alloys—theory and application to Cu–1.9 at% Ti and Ni–14 at% Al. In: Haasen P, Gerold V, Wagner R, Ashby MF (eds) Decomposition of alloys: the early stages. Pergamon, pp 91–103
Hou ZY, Babu RP, Hedström P, Odqvist J (2018) On coarsening of cementite during tempering of a martensitic steel, Unpublished 2018
Hillert M (1953) Nuclear composition—a factor of interest in nucleation. Acta Metall 1:764–766
Wu KS, Chen Q, Mason P (2018) Simulation of precipitation kinetics with non-spherical particles. J Phase Equilib Diffus 39:571–583
Kozeschnik E, Svoboda J, Fischer FD (2006) Shape factors in modeling of precipitation. Mater Sci Eng A 441:68–72
Acknowledgements
This work was performed within the VINN Excellence Center Hero-m, financed by VINNOVA, the Swedish Governmental Agency for Innovation Systems, Swedish industry and KTH Royal Institute of Technology. The authors are grateful to Fredrik Lindberg and Niklas Pettersson at Swerea KIMAB, Andrey Karasev and Hongying Du at KTH for electrolytic extraction assistance. The authors are also grateful for the constructive critic and comments on this manuscript from Dr. Manon Bonvalet.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that the contents have no conflict of interest toward any individual or organization.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hou, Z., Babu, R.P., Hedström, P. et al. Early stages of cementite precipitation during tempering of 1C–1Cr martensitic steel. J Mater Sci 54, 9222–9234 (2019). https://doi.org/10.1007/s10853-019-03530-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-019-03530-8