
Abstract
Crataegus scabrifolia is a significant botanical resource in Southwest China, renowned for its medicinal properties and high potential for development due to its rich medicinal components. However, genomic research on C. scabrifolia remains limited. This study conducted a comprehensive genome-wide survey of C. scabrifolia, employing flow cytometry in conjunction with genome K-mer analysis to assess its genomic characteristics in detail. Our findings reveal that despite a genome size similar to cultivated hawthorn (Crataegus pinnatifida var. major), C. scabrifolia exhibits a significantly lower heterozygosity rate of 0.5% compared to 1.77% observed in cultivated varieties. Additionally, we identified transposable elements comprising 51.79% of the assembled genome, with retrotransposons accounting for 35.05% of the total genome. Furthermore, this study identified numerous simple sequence repeat (SSR) marker loci and conducted a polymorphism analysis. Subsequently, we annotated the functions of single-copy genes, providing insights into the adaptive strategies and genetic stability of C. scabrifolia under varying environmental conditions. These findings offer crucial tools and resources for further genotype selection, genetic analysis, and breeding improvement.
Similar content being viewed by others

Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.Avoid common mistakes on your manuscript.
Introduction
Crataegus L., belonging to the Rosaceae family, subfamily Maloideae, is widely distributed throughout the northern hemisphere, with certain species being significant in both medicinal and culinary domains. In China, hawthorn boasts a long history of traditional medicinal and culinary use (Yang et al. 2022). Known for its sour–sweet taste, it is valued for its therapeutic effects including digestion aid, qi regulation, blood circulation improvement, and lipid reduction (Orhan 2018). Modern medical research has extensively documented hawthorn's pharmacological activities, particularly in cardiovascular health, encompassing cardiotonic, antiarrhythmic, hypotensive, hypolipidemic, and antioxidant properties. Presently, hawthorn is primarily employed in treating conditions such as congestive heart failure, hypertension, atherosclerosis, and digestive ailments (Chang et al. 2002).
The southwestern region of China benefits from unique geographical and climatic conditions, fostering exceptionally rich plant diversity and serving as a prominent area for hawthorn distribution (Zhang et al. 2014). Yunnan province, situated in southwestern China, stands out as a principal cultivation area for C. scabrifolia, renowned locally for its medicinal use. Research indicates that C. scabrifolia exhibits high levels of bioactive compounds and potent pharmacological effects. For instance, C. scabrifolia fruits demonstrate significant scavenging activity against superoxide anion radicals in vitro (Zhou et al. 1999). Moreover, the total flavonoid content in C. scabrifolia fruits (84 g/kg) surpasses that of cultivated hawthorn fruits (31 g/kg) by more than double (Gao and Feng 1994), underscoring its substantial medicinal value and developmental potential.
Currently, the most extensively studied and cultivated species of the Crataegus genus is the Crataegus pinnatifida var. major. However, research on the second most cultivated variety, the C. scabrifolia, is scarce and mainly focuses on the chemical composition of its fruits, their biological activity, and the evaluation of variety resources (Liu et al. 2010; Wu et al. 2014, 2023). Although some scholars have conducted chloroplast genomic research on C. scabrifolia (Wu et al. 2022), its genetic foundation remains underexplored. Genomic research can uncover the most comprehensive genetic variation sites in an individual, providing precise and extensive information for studying plant origin and evolution, gene function analysis, and genetic breeding. Microsatellite markers, that is, simple sequence repeats (SSR), developed based on the genome, are commonly used molecular markers in population genetics, gene targeting, and crop germplasm research. The identification of SSR loci can provide crucial data support for genetic improvement and breeding programs.
To elucidate the genomic characteristics and potential genetic resources of C. scabrifolia, enhancing our understanding and facilitating future molecular breeding efforts. We employ flow cytometry and K-mer analysis based on whole-genome sequencing technology to estimate the genome size of C. scabrifolia and analyze its genomic characteristics. Using bioinformatics methods, transposons across the whole genome were identified and classified, followed by the extraction of microsatellite sequence information. Additionally, we conducted functional annotation and analysis of single-copy genes in the genome, as well as the construction of the phylogenetic tree. These efforts aim to lay a solid foundation for further genomic analysis, gene discovery, and molecular genetic breeding.
Materials and methods
Plant and DNA sources
The experimental materials were sourced from the Kunming Institute of Botany, Chinese Academy of Sciences (25°14′ N, 102°75′ E, elevation 1900 m), utilizing fresh, tender leaves. A voucher specimen, C. scabrifolia (YUNCM5301260363), is deposited in the herbarium of Yunnan University of Traditional Chinese Medicine. Genomic DNA was extracted using the CTAB method (Doyle and Doyle 1987), and the DNA concentration and quality were assessed using a NanoDrop 2000 spectrophotometer and agarose gel electrophoresis.
Flow cytometry estimation of genome size
We selected Corn B73, sourced from the Kunming Institute of Botany, Chinese Academy of Sciences, as the internal reference.Samples were placed in MG dissociation solution, finely chopped vertically, and left to stand for 10 min. The nuclei were then filtered and treated with propidium iodide (PI) and RNAase solution, stained on ice in the dark for 1 h (Dolezel and Bartos 2005; Dolezel et al. 2007).
The stained nuclei suspension samples were mixed with the nuclei suspension of the reference sample. The fluorescence intensity of the stained nuclei suspension samples was measured using a BD FACScalibur (BD Biosciences, USA) flow cytometer. The coefficient of variation (CV) was controlled within 5%, and the data were analyzed using Modifit 3.0 software (Tian et al. 2011). The DNA content of the C. scabrifolia sample was calculated by comparing the relative fluorescence intensity between the corn and C. scabrifolia samples using the formula: DNA content of C. scabrifolia sample = DNA content of corn sample × fluorescence intensity of C. scabrifolia sample/fluorescence intensity of corn sample.
DNA sequencing, genome size and ploidy estimation
DNA samples were randomly sheared using an ultrasonic disruptor, targeting fragments approximately 350 bp in length. These fragments were then used to construct libraries for paired-end sequencing on the Illumina HiSeq platform. Sequencing data underwent quality assessment with Q20 and Q30 scores, and were subjected to filtering and quality control using fastp and FastQC software. Filtered data were further analyzed using a K-mer based approach (K = 19), and genome size, repetitive sequence content, and heterozygosity were estimated using findGSE (Sun et al. 2018) and genomescope (Ranallo-Benavidez et al. 2020). Ploidy estimation was performed using smudgeplot (Ranallo-Benavidez et al. 2020).
Genome assembly and GC content analysis
The high-quality filtered sequences were assembled using SOAPdenovo2 (Luo et al. 2012) with K-mer = 59 and default parameters, followed by the calculation of GC content. The completeness of the genome assembly and annotation was then assessed using Busco v5 (Simão et al. 2015). The genome sequence of C. scabrifolia was analyzed and compared against a single-copy ortholog database of embryophytes, resulting in the determination of the coverage rate of the C. scabrifolia genome against single-copy ortholog genes in the database.
Species matching analysis of C. scabrifolia using the NT database
To investigate the diversity of C. scabrifolia species and to assess whether the extracted sample DNA was contaminated, we conducted a comprehensive analysis using a constructed DNA library. From this library, we randomly selected 10,000 reads and performed BLAST (Basic Local Alignment Search Tool) comparisons against the NCBI nucleotide database (NT database). The origins of these sequences were determined based on their sequence similarity.
Identification and annotation of transposons
In the process of transposon identification and annotation, we utilized RepeatModeler v.1.08 (Abrusán et al. 2009) to construct a comprehensive de novo repeat library, employing its default settings for initial training. Following this, we applied RepeatMasker v4.0.7 (Bedell et al. 2000) to systematically identify and classify repetitive sequences within the genomic data. RepeatMasker enabled detailed annotation by comparing the sequences against the custom repeat library generated in the previous step.
SSR data mining and polymorphism analysis
We utilized SSRMMD software (Gou et al. 2020) to search for and enumerate SSR sites within the whole genome of C. scabrifolia. The criteria for counting were as follows: mononucleotide repeats were cataloged with a minimum of 10 consecutive nucleotides, dinucleotide repeats with at least 6 repeats, and trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide repeats were identified with a minimum of 5 repeats each. Additionally, for SSR comparison, we incorporated the cultivated hawthorn genome (https://www.rosaceae.org/) and conducted SSR locus searches using the same parameters. Subsequently, primers were designed using Primer3, and 12 randomly selected primers were employed to perform PCR amplification on C. scabrifolia samples collected from ten different regions in Yunnan. The PCR products were analyzed using capillary electrophoresis. GenAlEx 6.502 and Power Marker V3.25 were utilized to calculate various genetic parameters for C. scabrifolia, including the number of alleles (Na), the effective number of alleles (Ne), observed heterozygosity (Ho), expected heterozygosity (He), Shannon's information index (I), and polymorphism information content (PIC).
Gene annotation
Single-copy genes within C. scabrifolia were systematically identified using Busco v5 (Simão et al. 2015). Functional annotation of these genes was conducted using eggNOG-mapper v2.1.5 software (Cantalapiedra et al. 2021), leveraging the eggNOG database to annotate EuKaryotic Orthologous Groups (KOG), Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG).
Phylogenetic analysis
In this study, we employed identified single-copy homologous genes to construct a phylogenetic tree encompassing eight genera within the Rosaceae family. Subsequently, we applied the MCMCtree method, a sophisticated Bayesian framework available in the PAML 4.9 software package (Yang 2007).
Results
Genome size analysis of C. scabrifolia using flow cytometry
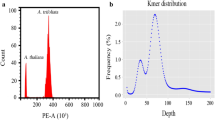
The genome size of C. scabrifolia was estimated using maize (genome size of 2.3 Gb) as a reference. The particle clusters were distinct, clear, and concentrated (Fig. 1a–c), indicating that the samples were well distinguished under the experimental conditions. The results showed that the fluorescence value for the maize internal standard peak was 61.85, while the fluorescence value for the C. scabrifolia peak was 22.76. The genome size of C. scabrifolia is 0.85 Gb, which is 0.37 times that of corn.

Genome size analysis. a Flow cytometry two-dimensional density map. b Flow cytometry two-dimensional dot plot. c Flow cytometry histogram. d K-mer frequency distribution (K = 19)
DNA sequencing and estimation of genome size
We sequenced the DNA of C. scabrifolia using the Illumina HiSeq PE platform, generating an initial dataset of 134 Gb. Following stringent quality control to remove low-quality reads, we secured 131 Gb of high-quality clean reads. The high fidelity of our sequencing is reflected in the Q20 and Q30 quality scores, which exceeded 96% and 90%, respectively, confirming the robustness of our sequencing data (Table 1).
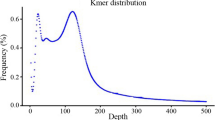
To estimate the genome size and ploidy of C. scabrifolia, we utilized K-mer analysis with K = 19. The resulting K-mer frequency distribution (Fig. 1d) revealed a genome size of approximately 0.87 Gb. This analysis also showed that the genome comprises about 57.86% repetitive sequences and exhibits a moderate heterozygosity rate of approximately 0.5% (Table 1). Initial ploidy estimation identified C. scabrifolia as an allotetraploid (AABB-type) (Fig. 2a). However, the K-mer distribution analysis suggests that the current genome structure is diploid, which may be the result of a relatively recent whole-genome duplication event.

Genomic characteristics. a Crataegus scabrifolia ploidy analysis. b BUSCO assessment. c Distribution of GC content and average sequencing depth. The x-axis represents K-mer depth (k = 27), while the y-axis represents the product of K-mer depth and GC content. d Transposon Distribution. The outermost circle represents the assembled genome, with tick marks every 25 Mb. The region from 0 to 580 Mb corresponds to scaffolds, and the region from 580 to 870 Mb corresponds to contigs. Tracks show, from outside to inside, the distribution of DNA transposons (red), LINE transposons (orange), LTR/Copia transposons (yellow), LTR/Gypsy transposons (blue), and SINE transposons (green)
Genome assembly and GC content analysis
We performed de novo genome assembly of C. scabrifolia using SOAPdenovo2 (K-mer = 59). This process yielded 3,516,690 raw contigs with a total length of 866,848,161 bp and a contig N50 of 306 bp. The final assembled genome comprised 2,361,006 scaffolds with a total length of 874,017,016 bp and a scaffold N50 of 3587 bp. The GC content of the assembled genome was 37.98% (Table 2).
Using BUSCO (Benchmarking Universal Single-Copy Orthologs) to evaluate the completeness of the genome assembly annotation, we found that 87.6% of the genes were covered, with 67.8% identified as single-copy genes and 19.8% as multi-copy genes (Fig. 2b).
We also performed a correlation analysis between K-mer depth and GC content in the sequencing results (Fig. 2c). The distribution shows no significant GC bias in sequencing, with sequencing depth primarily concentrated in the 50 ×–150 × range, indicating no contamination during sequencing. The average depth was 126 ×, with a minor sub-peak around 63 ×, which, when analyzed alongside Fig. 1d, may be attributed to heterozygosity within the genome.
Analysis of species matching in the C. scabrifolia NT database
To explore the genetic diversity of C. scabrifolia species and ensure the integrity of the extracted DNA samples, we conducted a comprehensive comparison of genomic fragment data. From the filtered high-quality data, we randomly selected 10,000 reads and subjected them to BLAST analysis against the NT database (Table 3).
The analysis revealed that Crataegus laevigata, had the highest match rate of 81.63% with the reads from the C. scabrifolia genome. This finding is significant, as both species belong to the Crataegus genus, indicating a close genetic relationship. Furthermore, other matching results included reads from various plants such as Malus domestica (apple), Pyrus communis (European pear), and Eriobotrya japonica (loquat), all of which are members of the Rosaceae family. This consistent match with other Rosaceae species supports the reliability of our genomic data. Importantly, our analysis did not identify any reads matching animal or microbial genomes, thus confirming the absence of contamination in our samples.
Identification and annotation of transposons
In our study of the C. scabrifolia genome, we identified a total of 2,361,006 transposon sequences, which collectively represent 51.79% of the entire genome. Among these transposons, long terminal repeat retrotransposons (LTRs) were the most prevalent, making up 35.05% of the genome. This predominance underscores the significant role LTRs play in shaping the genomic architecture and evolutionary history of the C. scabrifolia. On the other hand, DNA transposons were found to be much less common, constituting only 5.73% of the genome (Table 4). Additionally, we analyzed and presented the distribution patterns of the five major types of transposons within the genome (Fig. 2d). LTR transposons are more abundant in contigs compared to scaffolds, while SINE transposons display the opposite pattern, being more prevalent in scaffolds than in contigs.
SSR data mining and polymorphism analysis
In our de novo search for SSR loci within the C. scabrifolia genome, we identified a total of 493,829 SSR loci. The most common type was mononucleotide repeats (281,350 loci, 56.97%), followed by dinucleotide repeats (174,220 loci, 35.28%), trinucleotide repeats (31,646 loci, 6.41%), tetranucleotide repeats (3963 loci, 0.80%), pentanucleotide repeats (2069 loci, 0.42%), and hexanucleotide repeats (581 loci, 0.12%) (Fig. 3).

Quantities and distribution of different SSR types in C. scabrifolia. a Distribution of Various Repeat Types. b Repeat frequency of different mononucleotides. c Repeat frequency of different dinucleotides. d Repeat frequency of different trinucleotides
Similarly, SSR locus searches in cultivated hawthorn identified 402,799 SSR loci with a distribution pattern mirroring that of the C. scabrifolia. Mononucleotide repeats were predominant, comprising 55.54% (223,698 loci), followed by dinucleotide repeats with 35.92% (144,694 loci), and trinucleotide repeats with 6.74% (27,155 loci). Tetranucleotide repeats were present at 0.83% (3336 loci), pentanucleotide repeats at 0.77% (3114 loci), and hexanucleotide repeats at 0.20% (802 loci).
Twelve primer pairs were employed to analyze 10 C. scabrifolia samples (Table 5), resulting in the detection of 55 alleles across the loci. The number of alleles per locus ranged from 2 to 9, with an average of 4.583 alleles per locus. Particularly, locus C11 exhibited the highest number of alleles. The average effective number of alleles (Ne) was 3.273, with the highest value observed at the C11 locus (7.143). Observed heterozygosity (Ho) spanned from 0.1 to 0.8, while expected heterozygosity (He) ranged between 0.095 and 0.850. Shannon's information index (I) varied from 0.199 to 2.082, reflecting the genetic diversity across loci. The polymorphism information content (PIC) values ranged from 0.090 to 0.845, with an average value of 0.536, exceeding 0.5, indicating that the selected primer pairs demonstrated high levels of polymorphism.
Gene annotation
A total of 996 protein-coding genes in the KOG database have been annotated, accounting for 91.04% of the total predicted protein-coding genes. The KOG annotation statistics indicate that, excluding those with unknown functions (S), the most annotated COG function in the C. scabrifolia genome is replication, recombination, and repair (L), with 100 protein-coding genes, representing 10% of all protein-coding genes. The next most annotated functions are posttranslational modification, protein turnover, chaperones (O) with 86 genes (8.63%), translation, ribosomal structure, and biogenesis (J) with 79 genes (7.93%), RNA processing and modification (A) with 68 genes (6.83%), and carbohydrate transport and metabolism (G) with 43 genes (4.32%).
The Gene Ontology (GO) project was developed to address inconsistencies in gene function definitions across different databases and species, aiming to provide a unified and standardized gene function annotation system. In the C. scabrifolia genome, 627 genes are annotated in the GO database, which is 57.31% of the total predicted genes. Among the three major categories of the GO database—Molecular Function (MF), Biological Process (BP), and Cellular Components (CC)—the most enriched categories are cellular anatomical entity, intracellular, cellular process, metabolic process, and catalytic activity, with 545, 545, 518, 459, and 325 genes, respectively.
The Kyoto Encyclopedia of Genes and Genomes (KEGG) is one of the databases used to understand higher-order functional and biological systems such as cells, organisms, and ecosystems, as well as for studying pathways. In the C. scabrifolia genome, 301 genes are annotated in the KEGG database, accounting for 27.51% of the total predicted genes. The metabolism category has the highest number of enriched genes with 156. The next most enriched pathways are translation, replication and repair, and metabolism of cofactors and vitamins (Fig. 4).

Functional Classification of All Predicted Single-Copy Genes. a KOG Annotations of Predicted Single-Copy Genes. b GO Annotations of Predicted Single-Copy Genes. c KEGG Annotations of Predicted Single-Copy Genes
Phylogenetic analysis
To better understand the evolutionary relationships of C. scabrifolia, we constructed a phylogenetic tree including eight genera of the Rosaceae family. The results show that the divergence of the hawthorn genus from other Rosaceae genera occurred approximately 29.7 (27.9–32.3) million years ago (Mya), consistent with previous results (Zhang et al. 2022). Additionally, the divergence between C. scabrifolia and cultivated hawthorn was estimated to be around 16.3 (12.7–19.8) Mya (Fig. 5).

Phylogenetic tree of eight genera in the Rosaceae family. The numbers at the branch node in the tree indicate the divergence time and 95% confidence interval, the red text represents the species in this study
Discussion
Genome size, also known as DNA C-value, refers to the amount of DNA contained in a gamete of an organism. The C-value varies significantly among different species and can be used to assess biological characteristics of plants. In this study, we estimated the genome size of C. scabrifolia to be 870 Mb with a heterozygosity rate of 0.5% and a repeat sequence proportion of 57.86%, using flow cytometry combined with genome survey-based K-mer analysis. The genome size of cultivated hawthorn was determined to be 856.88 Mb, with a heterozygosity rate of 1.77% and a repeat sequence proportion of 67.89%. Although the genome sizes of the two hawthorn species are similar, the heterozygosity rate of C. scabrifolia is lower than that of cultivated hawthorn. This lower heterozygosity in C. scabrifolia may be attributed to its geographically restricted distribution, which limits gene flow between populations and results in lower genetic diversity (Favre et al. 2022). In contrast, cultivated hawthorn has a wider distribution and has undergone extensive artificial selection and hybridization to enhance yield, disease resistance, and adaptability, thus increasing its heterozygosity (Zhuang et al. 2022).
The higher proportion of repeat sequences in the cultivated hawthorn genome compared to the C. scabrifolia genome is likely due to the sequencing techniques used. Our study utilized only second-generation sequencing data, which has shorter read lengths and is less effective at accurately assembling highly repetitive genomic regions. Consequently, the identification of repeat sequences in the C. scabrifolia genome may be incomplete. In contrast, the genome of cultivated hawthorn was sequenced using a combination of second-generation and third-generation sequencing technologies, which provide longer read lengths that can span repetitive regions, resulting in a more comprehensive genome assembly. Thus, the lower proportion of detected repeat sequences in the C. scabrifolia genome can be attributed to the limitations of the sequencing technology employed in this study.
To further study the species diversity of C. scabrifolia, the top 10,000 reads from sequencing were extracted and aligned with the NT database using BLAST. The results indicated that among the published sequences, C. scabrifolia shares the highest read match rate with C. laevigata, indicating the closest phylogenetic relationship, followed by a closer relationship with apples.
Large-scale genome sequencing technology has provided new opportunities for the study of transposons. Research has shown that although the content of transposons varies among species, their content is closely related to genome size, showing a positive correlation. It is generally believed that genome size is influenced by both the increase and deletion of DNA content, with an increase in transposon copies being a significant factor in genome enlargement (Hawkins et al. 2008). Analysis of the C. scabrifolia genome revealed various types of transposons, among which LTRs are the predominant form, accounting for 51.79%. In contrast, DNA transposons are less common, making up only 5.73%. The high proportion of LTRs suggests that they may be involved in genome size expansion and functional diversification through mechanisms such as gene duplication, insertion mutations, and gene expression regulation (Vitte and Panaud 2003). Additionally, the abundance of LTR/Copia and LTR/Gypsy retrotransposons is significantly higher in contigs compared to scaffolds. This may be due to their longer sequences and the frequent presence of repetitive regions, which can complicate sequence assembly and lead to difficulties in joining these sequences accurately. Consequently, these transposons are more likely to appear in shorter, more fragmented contigs, whereas their presence in longer scaffolds is reduced or less complete due to the increased assembly difficulty. On the other hand, SINE transposons, characterized by their shorter sequences and lack of extensive repetitive regions, are generally more easily recognized and assembled during genome assembly. As a result, SINE transposons are more commonly found in longer scaffolds, where they can be accurately integrated into the genome assembly (Treangen and Salzberg 2012).
Understanding the diverse roles and impacts of these transposons is crucial for elucidating the genetic and evolutionary processes that shape the genome of C. scabrifolia.
Breeding research on C. scabrifolia has been relatively limited, but molecular marker technology can facilitate the selection and preservation of genotypes. Genomic SSR markers are highly polymorphic molecular markers known for their co-dominant inheritance, excellent reproducibility, and stability. These characteristics make SSR markers highly valuable for genomic research, genetic analysis, and breeding improvement (Lei et al. 2021). Due to the lack of genomic data specific to Crataegus species, previous research utilized SSR loci developed from apple and pear genomes to analyze various hawthorn genotypes (Güney et al. 2018). In this study, we identified 493,829 SSR loci in the C. scabrifolia genome and conducted polymorphism analysis and evaluation, The results showed that these SSR loci exhibit relatively high levels of polymorphism within the species, providing a comprehensive molecular marker resource for genotype selection and preservation. These newly identified markers are expected to significantly enhance the precision and effectiveness of related research. The observed differences in SSR sequence abundance between C. scabrifolia and cultivated hawthorn could be attributed to selective pressures or genetic drift (Bagshaw 2017). Investigating the genomic and evolutionary mechanisms underlying these differences will offer valuable insights into the genetic diversity and adaptability of hawthorn species.
Single-copy genes, due to their high conservation and functional importance, exhibit unique value in phylogenetic analysis and gene function research (Li et al. 2003). These genes are typically unique within the genome and are integral to fundamental biological processes. They maintain a high degree of conservation across species evolution, reflecting their essential roles. In our study, we conducted a comprehensive search for single-copy genes within the genome of C. scabrifolia and subsequently annotated them using the KOG, GO, and KEGG databases. The categorization of these annotated genes revealed their involvement in crucial biological functions, with significant classifications including “Replication, Recombination, and Repair,” “Cellular Organization and Structure,” and “Metabolism.” These genes are integral to maintaining genomic stability, ensuring cellular structural complexity, and regulating metabolic processes. This underscores the adaptive strengths of C. scabrifolia in varied and stressful environments, potentially aiding in its survival and reproduction under adverse conditions (Chinnusamy and Zhu 2009). Detailed analysis of these genes' expression patterns can offer significant insights into their dynamic regulatory mechanisms across different environmental contexts. Such insights are crucial for advancing genetic improvement, informing conservation strategies, and supporting further functional genomics research.
Additionally, we utilized the constructed phylogenetic tree to gain insights into the evolutionary relationships among different species. This analysis revealed the divergence time between C. scabrifolia and cultivated hawthorn, which is significant for studying the evolutionary history and genetic diversity of these species.
Conclusion
This research, through the inaugural comprehensive analysis of the genomic characteristics and pivotal genetic elements of C. scabrifolia, unveils significant advantages related to genome stability and adaptability. It provides new insights into its genetic evolution and environmental adaptation mechanisms and offers valuable theoretical foundations for future functional genomics research, molecular breeding, and conservation management.
References
Abrusán G, Grundmann N, DeMester L, Makalowski W (2009) TEclass–a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 25:1329–1330. https://doi.org/10.1093/bioinformatics/btp084
Bagshaw ATM (2017) Functional mechanisms of microsatellite DNA in eukaryotic genomes. Genome Biol Evol 9:2428–2443. https://doi.org/10.1093/gbe/evx164
Bedell JA, Korf I, Gish W (2000) MaskerAid: a performance enhancement to RepeatMasker. Bioinformatics 16:1040–1041. https://doi.org/10.1093/bioinformatics/16.11.1040
Cantalapiedra CP, Hernández-Plaza A, Letunic I et al (2021) eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol Biol Evol 38:5825–5829. https://doi.org/10.1093/molbev/msab293
Chang Q, Zuo Z, Harrison F, Chow MS (2002) Hawthorn. J Clin Pharmacol 42:605–612. https://doi.org/10.1177/00970002042006003
Chinnusamy V, Zhu JK (2009) Epigenetic regulation of stress responses in plants. Curr Opin Plant Biol 12:133–139. https://doi.org/10.1016/j.pbi.2008.12.006
Dolezel J, Bartos J (2005) Plant DNA flow cytometry and estimation of nuclear genome size. Ann Bot 95:99–110. https://doi.org/10.1093/aob/mci005
Dolezel J, Greilhuber J, Suda J (2007) Estimation of nuclear DNA content in plants using flow cytometry. Nat Protoc 2:2233–2244. https://doi.org/10.1038/nprot.2007.310
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemistry 19:11–15
Favre F, Jourda C, Grisoni M et al (2022) A genome-wide assessment of the genetic diversity, evolution and relationships with allied species of the clonally propagated crop Vanilla planifolia Jacks. ex Andrews. Genet Resour Crop Evol 69:2125–2139. https://doi.org/10.1007/s10722-022-01362-1
Gao GY, Feng YX (1994) Pharmacognocy and resource utilization of Yunnan-Hawthorn. Chin Pharm J 06:329–331
Gou X, Shi H, Yu S et al (2020) SSRMMD: a rapid and accurate algorithm for mining SSR feature loci and candidate polymorphic SSRs based on assembled sequences. Front Genet 11:706. https://doi.org/10.3389/fgene.2020.00706
Güney M, Kafkas S, Keles H et al (2018) Characterization of hawthorn (Crataegus spp.) genotypes by SSR markers. Physiol Mol Biol Plants 24:1221–1230. https://doi.org/10.1007/s12298-018-0604-6
Hawkins JS, Grover CE, Wendel JF (2008) Repeated big bangs and the expanding universe: directionality in plant genome size evolution. Plant Sci 174:557–562. https://doi.org/10.1016/j.plantsci.2008.03.015
Lei Y, Zhou Y, Price M, Song Z (2021) Genome-wide characterization of microsatellite DNA in fishes: survey and analysis of their abundance and frequency in genome-specific regions. BMC Genom 22:421. https://doi.org/10.1186/s12864-021-07752-6
Li L, Stoeckert CJ Jr, Roos DS (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13:2178–2189. https://doi.org/10.1101/gr.1224503
Liu P, Kallio H, Lü D et al (2010) Acids, sugars, and sugar alcohols in Chinese hawthorn (Crataegus spp.) fruits. J Agric Food Chem 58:1012–1019. https://doi.org/10.1021/jf902773v
Luo R, Liu B, Xie Y et al (2012) SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1:18. https://doi.org/10.1186/2047-217X-1-18
Orhan IE (2018) Phytochemical and pharmacological activity profile of Crataegus oxyacantha L. (Hawthorn)—a cardiotonic herb. Curr Med Chem 25:4854–4865. https://doi.org/10.2174/0929867323666160919095519
Ranallo-Benavidez TR, Jaron KS, Schatz MC (2020) GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun 11:1432. https://doi.org/10.1038/s41467-020-14998-3
Simão FA, Waterhouse RM, Ioannidis P et al (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212. https://doi.org/10.1093/bioinformatics/btv351
Sun H, Ding J, Piednoël M, Schneeberger K (2018) findGSE: estimating genome size variation within human and Arabidopsis using k-mer frequencies. Bioinformatics 34:550–557. https://doi.org/10.1093/bioinformatics/btx637
Tian XM, Zhou XY, Gong N (2011) Applications of flow cytometry in plant research-analysis of nuclear DNA content and ploidy level in plant cells. Chin Agric Sci Bull 27:21–27
Treangen TJ, Salzberg SL (2012) Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet 13:36–46. https://doi.org/10.1038/nrg3117
Vitte C, Panaud O (2003) Formation of solo-LTRs through unequal homologous recombination counterbalances amplifications of LTR retrotransposons in rice Oryza sativa L. Mol Biol Evol 20:528–540. https://doi.org/10.1093/molbev/msg055
Wu J, Peng W, Qin R, Zhou H (2014) Crataegus pinnatifida: chemical constituents, pharmacology, and potential applications. Molecules 19:1685–1712. https://doi.org/10.3390/molecules19021685
Wu X, Luo D, Zhang Y et al (2022) Comparative genomic and phylogenetic analysis of chloroplast genomes of hawthorn (Crataegus spp.) in Southwest China. Front Genet 13:900357. https://doi.org/10.3389/fgene.2022.900357
Wu X, Luo D, Zhang Y et al (2023) Integrative analysis of the metabolome and transcriptome reveals the potential mechanism of fruit flavor formation in wild hawthorn (Crataegus chungtienensis). Plant Divers 45:590–600. https://doi.org/10.1016/j.pld.2023.02.001
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591. https://doi.org/10.1093/molbev/msm088
Yang YC, Wang EH, Wang JQ et al (2022) History of traditional Chinese medicine Crataegi fructus. Asia-Pacific Tradit Med 18:157–163
Zhang HP, Zhang JY, Liu QL (2014) Research progress on hawthorn germplasm resources and breeding varieties in China. China Seed Industry 02:15–17
Zhang T, Qiao Q, Du X et al (2022) Cultivated hawthorn (Crataegus pinnatifida var. major) genome sheds light on the evolution of Maleae (apple tribe). J Integr Plant Biol 64:1487–1501. https://doi.org/10.1111/jipb.13318
Zhou QP, Wang LW, Gao GY (1999) Stuty on antioxidative and decreasing blood-fat effect in four kinds of fructus Crataegi. Res Pract Chin Med 03:3–5
Zhuang Y, Li X, Hu J et al (2022) Expanding the gene pool for soybean improvement with its wild relatives. aBIOTECH 3:115–125. https://doi.org/10.1007/s42994-022-00072-7
Acknowledgements
The authors appreciate the bioinformatics high-performance computing server at Yunnan University of Chinese Medicine for providing computational resources for data analysis. This research was jointly funded by the National Natural Science Foundation of China (32260094), the Yunnan Provincial Traditional Chinese Medicine Joint Key Project (202101AZ070001-166), and the Yunnan Provincial Major Science and Technology Special Project (202102AE090031).
Funding
Yunnan Provincial Science and Technology Department, 202102AE090031, 202101AZ070001-166, The National Natural Science Foundation of China, 32260094.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval
Not applicable.
Consent for publication
All authors participated in, read and approved the final version of the article before publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, B., Wu, X., Luo, D. et al. Genome-wide survey of Crataegus scabrifolia provides new insights into its genetic evolution and adaptation mechanisms. Genet Resour Crop Evol 72, 3919–3932 (2025). https://doi.org/10.1007/s10722-024-02186-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-024-02186-x
Keywords
Profiles
- Ticao Zhang View author profile