
Abstract
The European whitefish Coregonus lavaretus complex represents one of the most diverse radiations within salmonids, with extreme morphological and genetic differentiation across its range. Such variation has led to the assignment of many populations to separate species. In Great Britain, the seven native populations of C. lavaretus (two in Scotland, four in England, one in Wales) were previously classified into three species, and recent taxonomic revision resurrected the previous nomenclature. Here we used a dataset of 15 microsatellites to: (1) investigate the genetic diversity of British populations, (2) assess the level of population structure and the relationships between British populations. Genetic diversity was highest in Welsh (HO = 0.50, AR = 5.29), intermediate in English (HO = 0.41–0.50, AR = 2.83–3.88), and lowest in Scottish populations (HO = 0.28–0.35, AR = 2.56–3.04). Population structure analyses indicated high genetic differentiation (global FST = 0.388) between all populations but for the two Scottish populations (FST = 0.063) and two English populations (FST = 0.038). Principal component analysis and molecular ANOVA revealed separation between Scottish, English, and Welsh populations, with the Scottish populations being the most diverged. We argue that the data presented here are not sufficient to support a separation of the British European whitefish populations into three separate species, but support the delineation of different ESUs for these populations.
Similar content being viewed by others


Introduction
Accurate species delineation is fundamental for the practical protection and conservation of biota (Mace 2004), but the use of genetic markers sometimes disagree with traditional morphology-based taxonomy (Bickford et al. 2007; Padial et al. 2010; Mayr 2011). Conflicts between morphological and molecular data are common in temperate freshwater fishes, as populations tend to be isolated from each other due to physical barriers, present a disjunct distribution, and show high levels of phenotypic and ecological differentiation (Taylor 1999; Adams et al. 2016; Moccetti et al. 2019).
The salmonid European whitefish Coregonus lavaretus forms a large species complex spanning across the north temperate regions of the Palaearctic, displaying high levels of phenotypic variation at the inter- and intra-population levels (e.g. Østbye et al. 2005a, b; Præbel et al. 2013a; Siwertsson et al. 2013; Ozerov et al. 2015; Adams et al. 2016) and rapid evolution under changing environmental conditions (Hudson et al. 2013; Jacobs et al. 2019). From an evolutionary perspective this species is a renowned example of repeated evolution across limnetic-benthic gradients in lacustrine environments (e.g. Østbye et al. 2005a, b; Vonlanthen et al. 2009; Præbel et al. 2013a; Siwertsson et al. 2013; Hudson et al. 2017), but for fish biologists it represents a taxonomic challenge, as morphological similarities do not necessarily reflect shared genetic history (e.g. Præbel et al. 2013a). This is due to the long-standing use of certain morphological, ecological, and meristic traits that might be environmentally influenced to discriminate coregonids into different species (Etheridge et al. 2012a). The number of gill rakers on the anterior gill arch is one the main characters used (Svärdson 1952, 1979; Kottelat and Freyhof 2007), which while genetically based, has been shown to be influenced by plasticity (Lindsey 1981) and under natural selection (Ozerov et al. 2015; Häkli et al. 2018; Jacobs et al. 2019) given its important role in feeding ecology (Kahilainen et al. 2011).
European whitefish is native to seven lakes in Great Britain, clustered into three distinct geographic areas. These are lochs Eck and Lomond in west-central Scotland, where it is known as powan, Red Tarn, Haweswater, Brotherswater, and Ullswater in north-west England, known as schelly, and Llyn Tegid in north Wales, known as gwyniad (Maitland and Campbell 1992) (hereafter referred to as Scottish, English and Welsh populations respectively). Apart from the lakes of Red Tarn and Brotherswater which are connected to Ullswater through small waterways, all lakes are isolated from one another. Originally, these populations were classified as three distinct species: populations in Scotland were described as Coregonus clupeoides Lacépède 1803, populations in England were classified as Coregonus stigmaticus Regan 1908, and the single Welsh population was classified as Coregonus pennantii Valeciennes 1848. Subsequently, these populations were recognised to be part of the Coregonus lavaretus species complex and grouped into a single species (Maitland and Campbell 1992). More recently, however, Kottelat and Freyhof (2007), largely on the basis of morphological characteristics, re-classified the entire European whitefish complex recognising 59 separate species across Europe and suggesting that there are many more waiting for formal description. For the United Kingdom (UK), Kottelat and Freyhof (2007) using morphological and meristic traits, including gill raker number, reinstated the late nineteenth century species classification, splitting the seven populations into three species (C. clupeoides, C. stigmaticus, C. pennantii).
The validity of the species boundaries and the identification key by Kottelat and Freyhof (2007) for the European whitefish populations in Britain was tested by Etheridge et al. (2012a). Whilst there exists an overall morphological separation between the Scottish, English, and Welsh populations, Etheridge et al. (2012a) concluded that the observed differences between them were insufficient evidence to split these populations into three separate species. Additionally, a large overlap existed between key morphological traits of putative different species, leading Etheridge et al. (2012a) to attribute the morphological differences between groups to phenotypic plasticity, genetic drift, and/or local adaptation. However, their study did not investigate to what extent the morphological separation was supported by genetic differentiation. Phenotypic and genetic information combined is necessary to gain a better understanding of the diversity present within populations, and to assess their conservation status. In particular, assessment of genetic variation and population structure forms the basis for the delineation of evolutionarily significant units (ESUs) (Quintela et al. 2010), a concept proposed by Ryder (1986) which might be more fitting to the scenario of many freshwater fish species whose high diversity is associated with post-glacial recolonization of northern Europe (April et al. 2013). Therefore, an accurate assessment of the genetic population structure of the European whitefish populations in Britain is needed, in particular with markers such as microsatellites that have been shown to be useful in the delimitation of ESUs (e.g. Mockford et al. 2006; de Oliveira et al. 2008; Zhu et al. 2013). Developing an understanding of the genetic diversity within and between populations, in addition to phenotypic variation, that exists within these populations is of significant importance if an adequate conservation strategy for this species is to be achieved (Brodersen and Seehausen 2014).
To date, genetic research on C. lavaretus in the UK has been limited and there is no clear consensus on the relationship between populations. Early studies using electrophoretic analysis of proteins on C. lavaretus and the two other Coregonus species currently recognised in the British Isles C. albula, and C. autumnalis, (Maitland and Campbell 1992), showed that the seven C. lavaretus populations constitute a single species distinct from the other two species (Ferguson 1974). A later study, using allozymes, focused on the English and Welsh populations of C. lavaretus showing the Welsh population to be nested within the English group (Beaumont et al. 1995). However, mitochondrial restriction fragment length polymorphism (RFLPs) from another study showed the English populations to be monophyletic, and supported a closer relationship between the Scottish and Welsh populations (Hartley 1995).
European whitefish in the UK is a priority species for the UK Biodiversity Action Plan, and it is protected under Schedule 5 of the Wildlife and Countryside Act 1981 due to its rarity and vulnerability to pressures such as invasive species, water quality and habitat degradation (Maitland and Lyle 2013; Winfield et al. 2013). This has led to the establishment of several translocated populations using stocks from Scotland (Maitland and Lyle 2013; Adams et al. 2014), England (Winfield et al. 2013), and Wales (Thomas et al. 2013), as a means of mitigating potential threats (Præbel et al. 2019). Therefore a better understanding of genetic diversity and structuring is of primary importance for the appropriate conservation of European whitefish in Britain.
In the present study, we applied a population genetics analysis of all native European whitefish populations in Britain to: 1) assess their overall genetic diversity and compare it to published data on other European populations; and 2) determine the level of genetic structuring between populations. The outputs of the study are then viewed in a conservation policy context.
Methods
Sampling
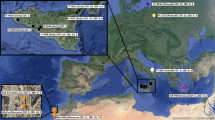
Sampling was conducted between 2005 and 2009. Samples were collected from six native populations in Britain (Fig. 1); Loch Lomond (LLA, n = 41) and Loch Eck (ECK, n = 36) in Scotland, Llyn Tegid (LTE, n = 44) in Wales, Red Tarn (RTA, n = 22), Brotherswater (BWA, n = 19) and Ullswater (UWA, n = 29) in the Lake District of north-west England. Samples from Haweswater (HAW, n = 46) were collected in 1986. Samples were collected using Norden benthic gill nets as described in Etheridge et al. (2012a). Details about lake characteristics are reported in Table S1.

Locations of Loch Eck, Loch Lomond, Llyn Tegid and the English Lake District (containing Brotherswater, Haweswater, Red Tarn and Ullswater) in the U.K. Note that all locations except Haweswater and Ullswater are not visible at the scales of the figure. Partly redrawn with permission from Ramsbottom (1976). The three geographic clusters of populations are shown. Lochs Eck and Lomond are in west-central Scotland (SCT), Llyn Tegid in north central Wales (WLS), and the other four lakes in the north west of England (ENG)
Laboratory procedures
Genomic DNA was extracted from gill filaments, fin clips or scales using E-Z96 Tissue DNA Kit (OMEGA Bio-tek) following the manufacturer instructions. A total of 15 microsatellite loci (Table S2, Patton et al. 1997; Susnik et al. 1999; Turgeon et al. 1999; Rogers et al. 2004; Winkler and Weiss 2008) were amplified in four polymerase chain reaction (PCR) multiplexes in 2.5 µl reactions following the protocol by Præbel et al. (2013b). Briefly, the PCR profiles for all multiplex assays (mplx) consisted of an initial denaturation step at 95 °C for 15 min and a final elongation step of 30 min at 60 °C. The amplification cycles consisted of: mplx (I) 25 cycles of 95 °C for 30 s, 57 °C for 3 min, and 72 °C for 1 min; mplx (II) 25 cycles of 95 °C for 30 s, 60 °C for 3 min, and 72 °C for 1 min; mplx (III) 26 cycles of 95 °C for 30 s, 61 °C for 3 min, and 72 °C for 1 min; mplx (IV) 27 cycles of 95 °C for 30 s, 60 °C for 3 min, and 72 °C for 1 min. The fluorescent-labeled PCR products were separated on an ABI 3130 XL Automated Genetic Analyzer (Applied Biosystems) and sized according to the GeneScan LIZ 500 internal size standard (Applied Biosystems).
Genotyping, validation and quality control of genotypic data
Alleles were automatically binned in predefined allelic bins by the GeneMapper 3.7 software (Applied Biosystems) and verified twice by visual inspection. After the first validation of the genotypes, 3–9% of individuals per population were re-extracted and re-run at all 15 loci to ensure properly amplified alleles, i.e. reducing the possibility for methodological errors such as large allele drop-out and stutter scoring. The genotypes resulting from the initial run and the rerun were manually compared for all individuals to rule out miss-scoring of alleles. If any doubt occurred in this comparison the sample was re-extracted and re-run at all loci to obtain a consensus genotype. No mismatch was identified in the dataset. The samples were finally screened for abnormalities (null alleles, scoring errors, etc.) in the software MICRO-CHECKER 2.2.3 (Van Oosterhout et al. 2004), using 1,000 bootstrap iterations over loci to generate the expected homozygote and heterozygote frequencies.
Genetic diversity and variation
Expected and observed heterozygosity, allele and effective allele numbers, and FIS were calculated in GenoDive v. 2.0b27 (Meirmans and Van Tienderen 2004) per population and per locus. Allelic and private allelic richness per population and per locus were determined using a rarefaction procedure, to account for sample size differences, for the smallest sample size (38 genes) implemented in HP-RARE v. 1.0 (Kalinowski 2005). Tests for linkage disequilibrium (LD) between pairs of loci were conducted in the R package Genepop v. 1.0.5 (Rousset 2008) with the following settings: 100,000 dememorization, 1000 batches, 10,000 iterations. Deviations from Hardy–Weinberg equilibrium (HWE) for loci within populations were tested in GenoDive using 1000 permutations of alleles among individuals. Pairwise p-values from the LD and HWE tests were corrected for multiple comparisons by sequential Bonferroni corrections (Rice, 1989). We also tested for a correlation between latitude and observed heterozygosity and allelic richness using Spearman correlation in the R environment (R Core Team 2019).
Population structure
FST (Weir and Cockerham 1984) was calculated at the global level and pairwise between populations using the R package hierfstat v. 0.04-22 (Goudet 2005) and GenoDive respectively, and tested for significance using 10,000 permutations of genotypes among populations.
Discriminant analysis of principal components (DAPC) (Jombart et al. 2010) implemented in the R package adegenet v.2.1.1 (Jombart 2008) was used to identify clusters of genetically related individuals. DAPC is a method that relies on transforming the allele frequency data into principal components that are then passed onto the discriminant analysis (DA), which maximises variation between groups and minimises it within groups (Jombart et al. 2010). The DAPC method works in two ways: (1) if prior knowledge of the genetic groups that individuals belong to exists, then these genetic groups will be used as priors in the analysis for assignment to group membership; (2) when no such information exists, genetic groups can be identified using k-means clustering sequentially with increasing values of k, with the best k value being the one scoring the lowest Bayesian information criterion (BIC), and individuals assigned to such groups. We ran the DAPC both with lake of origin as prior information for group membership, and without prior information (testing k values from 1 to 10), and compared the results to assess if populations show evidence of structure by lake. We then visualised the genetic clusters using the complot function in adegenet, which produces a barplot of group assignment probabilities, and using scatterplots. The xvalDAPC function in adegenet was used to determine the number of principal components (PCs) to be retained by the DAPC analysis.
In addition to DAPC, which maximises between-group variation, we also ran a principal component analysis (PCA), which summarise overall genetic variation, in adegenet. For the DAPC and PCA analyses, 15 individuals (two from Lomond, Eck and Ullswater, three from Llyn Tegid, and six from Haweswater) that had missing data at one or more loci were excluded.
Analysis of molecular variance (AMOVA) using FST was employed to test the effect of hierarchical groupings on the genetic variance, by testing north (Lomond, Eck) and south (Llyn Tegid, Haweswater, Brotherswater, Ullswater, Red Tarn) grouping, and by geographic region (Lomond, Eck), (Llyn Tegid), (Haweswater, Brotherswater, Ullswater, Red Tarn), following the Kottelat and Freyhoff (2007) taxonomic designation. AMOVA was carried out using the R package poppr v.2.8.1 (Kamvar et al. 2014), using 1000 permutations of individuals among groupings to assess significant difference from a null hypothesis of panmixia.
A distance-based phylogenetic relationship among all the populations was constructed using unrooted Neighbor-joining (NJ) clustering analysis of Cavalli-Sforza and Edwards (1967) Chords genetic distances (Table 2). The tree was constructed in poppr with 1,000 bootstrap replicates with the function aboot and was rooted using mid-point rooting due to lack of a specific outgroup.
All the programs used for the genetic analyses, apart from HP-RARE and GenoDive, are part of the R environment (R Core Team 2019).
Results
Genotyping, validation and quality control of genotypic data
We did not identify any mismatch between the original individual multi-locus genotypes and the re-extracted replicates within the present dataset. Heterozygote deficits were indicated by MICRO-CHECKER at four loci in three populations (BWF2 (Ullswater), Cla-Tet1 (Llyn Tegid), Cla-Tet13 (Haweswater, Llyn Tegid), and Cla-Tet18 (Haweswater)). In two out of the four cases, the heterozygote deficit appeared to reflect the presence of null alleles. In the remaining two cases the heterozygote deficit was also associated with more than 50% of the alleles being in one allele class but seemed to result from stuttering. As this effect is not pervasive in the dataset, we suggest that the heterozygote deficit reflects genetic effects associated with the population history, e.g. low diversity, rather than the occurrence of null alleles and other locus specific abnormalities (but see discussion). This is further supported by the careful validation and quality control of the genotypic data where all poorly amplified samples were re-extracted. Consequently, all loci were retained in further analysis.
Genetic diversity and variation
Summary statistics for the microsatellite loci are reported in Table S3. The number of alleles per locus ranged from three to 29, with an average of 9.6 alleles per locus. Observed heterozygosity ranged from 0.17 to 0.72, with an average of 0.42 across loci with all populations combined. Six loci (BWF1, BWF2, Cocl_lav04, Cocl-lav10, BFRO_018, and Cocl_lav49) were monomorphic in one or several populations (Table S3). Deviations from linkage equilibrium were identified for five of 105 locus pair tests but all were non-significant after sequential Bonferroni corrections. Deviations from HWE were found in four populations at four loci, for a total of eight comparisons, but were not significant after Bonferroni corrections.
The Scottish populations showed slightly lower genetic variation and allelic richness compared to the populations from Wales and England (Table 1), with the Scottish population of Loch Lomond (HO = 0.35, AR = 3.04) having higher diversity than Loch Eck (HO = 0.28, AR = 2.56). Llyn Tegid in Wales was the most genetically diverse population (HO = 0.5, AR = 5.29), with the highest heterozygosity, allelic and private allelic richness (Table 1). Among the English populations, Haweswater had the highest heterozygosity and allelic richness (HO = 0.5, AR = 3.88) (Table 1) followed by Red Tarn (HO = 0.44, AR = 3.27), Brotherswater (HO = 0.46, AR = 3.00), and Ullswater (HO = 0.41, AR = 2.83). We found a significant negative correlation between latitude and observed heterozygosity (Spearman’s ρ = -0.96, p-value = 0.003), but no correlation between latitude and allelic richness (Spearman’s ρ = − 0.71, p-value = 0.088).
Population structure
Mean global FST was 0.388 (95% CI 0.308–0.478), and all pairwise FST values between populations were significant after 10,000 permutations (Table 2). The populations in Scotland showed high FST differentiation from the rest of the British populations, with values between 0.44 and 0.58. The lowest differentiation was observed between the English populations of Brotherswater and Ullswater (FST = 0.038), followed by the Scottish populations of Lomond and Eck (FST = 0.063). Genetic differentiation for the remaining English populations ranged from FST of 0.120 between Haweswater and Ullswater to FST of 0.290 between Red Tarn and Brotherswater. Genetic differentiation between Llyn Tegid and the English populations ranged from a FST value of 0.260 to a FST value of 0.315.
The DAPC without prior information identified six genetic clusters as most likely (Fig. 2a), after retaining 20 PCs as suggested by the xvalDAPC function. Individuals from Loch Lomond and Loch Eck were assigned to two separate clusters, although few individuals of each lake were assigned to the opposite cluster; in contrast, Llyn Tegid, Red Tarn, and Haweswater individuals formed separate clusters which did not include individuals originating from another site. Brotherswater and Ullswater samples were assigned to cluster 6 (Fig. 2a). The two clusters formed by individuals from Lomond and Eck were separated from the others along discriminant function 1 (DF1), which explained 72.7% of the variation, while DF2 explained 16.3% of the total variation (Fig. 2b), and separated Llyn Tegid from the English populations of Haweswater, Brotherswater and Ullswater, with Red Tarn in an intermediate distance between these two groups.

Analyses of population structure using DAPC and PCA. a Barplot showing assignment of individuals to the six clusters recovered by the DAPC without prior information. b Scatterplot of the DAPC without prior information, displaying clusters 1–6 along discriminant function (DF) 1 and 2. c Barplot showing the assignment of individuals to lakes of origin as recovered by the DAPC with prior information; d Scatterplot of the DAPC with prior information, displaying the genetic clusters corresponding to lakes along DF1 and DF2. Ellipses on 2B and 2D are inertia ellipses. E) Results of the principal component analysis, displaying PC1, PC2, and PC3. Colour codes are the same throughout the figure components
The DAPC with prior information had an overall proportion of correct assignment of individuals to populations (lake) of origin of 95.5%. Five individuals could not be assigned to either Lomond or Eck with more than 80% accuracy, and five individuals could not be assigned to either Brotherswater or Ullswater with more than 80% accuracy (Fig. 2c). DF1 explained 71.6% of the total variation, and separated Lomond and Eck from the other populations; DF2 explained 16.8% of the total variance and separated the remaining populations in a similar fashion as the DAPC without prior information (Fig. 2d).
Similar results were recovered by the PCA (Fig. 2e). The Scottish populations were separated from the rest along PC1, which explained 12.4% of the total variance, while PC2 separated the Welsh population from the English ones, explaining 6.6% of the variance. PC3, which explained 2.9% of the variance, separated Red Tarn and Haweswater from Brotherswater and Ullswater, and PC4, with 2.6% of the variance, separated Haweswater from Red Tarn (not shown).
We tested two hierarchical levels in the AMOVA (Table 3), to assess how variation was partitioned across the landscape. In the north-to-south grouping (Scotland)(Wales,England), variation between groups and variation between lakes within groups explained 33% and 14% of the total variance respectively, while in the grouping based on geographic origin (Scotland), (Wales), (England), variation between groups and variation between lakes within groups explained 35% and 9% of the total variance respectively, indicating the latter grouping as a more realistic representation of the landscape wide grouping.
In a mid-point rooted NJ tree, Lomond and Eck formed a cluster sister to all the other populations, and Llyn Tegid was then separated as a sister to a clade of all English populations (Fig. 3).

Neighbor-Joining tree calculated with Cavalli-Sforza and Edwards chord distances for the seven population of European whitefish in Britain. Mid-point rooting was applied to due to lack of outgroup. Support was calculated using 1000 bootstrap replicates
Discussion
Here we describe microsatellite genetic diversity and population structure in the seven native populations of European whitefish Coregonus lavaretus in Britain. These populations are of high conservation concern (Winfield et al. 2013), and our results provide important information for future conservation plans. We find a latitudinal gradient of genetic diversity, with the Welsh population having the highest genetic diversity and the Scottish populations the lowest. We also found evidence of population structuring, both between and within geographic areas.
Genetic diversity of British populations
Although comparison of genetic diversity estimates obtained from different microsatellite loci can only be qualitative, our findings suggest that the European whitefish populations in Britain, with Scottish ones in particular, tend to harbour lower genetic diversity compared to other European populations. Populations in Scotland had the highest number of monomorphic loci, with Loch Lomond and Loch Eck having three and four monomorphic loci respectively, Llyn Tegid, Red Tarn, and Ullswater had one monomorphic locus each, and Haweswater and Brotherswater had none despite having smaller sample sizes than Loch Lomond and Loch Eck. As a point of comparison, Häkli et al. (2018) sampled 27 European whitefish populations from Fennoscandia using 21 microsatellites, including the 15 we used, and found only five monomorphic loci. Furthermore, no monomorphic loci were observed in a dataset of 15 populations and 11 microsatellites, including eight loci present in our dataset, from Denmark (Hansen et al. 2008). Allelic richness across all British populations for 11 loci out of 15 analysed in this study was also lower (AR = 1.0–5.8) compared to whitefish populations from Fennoscandia (AR = 2.0–13.6) (Säisä et al. 2008; Præbel et al. 2013a; Häkli et al. 2018). Heterozygosity in the British populations ranged from 0.28 in Eck to 0.50 in Llyn Tegid, but a study looking at European whitefish in the Baltic region (Estonia to Sweden) with 12 microsatellites, seven of which overlapped with ours, found heterozygosity levels between 0.64 and 0.77 (Wennerström et al. 2013). Another study based on 529 individuals and 11 microsatellites, six of which overlapped with ours, found heterozygosity between 0.68 and 0.80 (Ozerov et al. 2015). A more similar level of heterozygosity, 0.47 to 0.58, was found in Alpine populations of whitefish (Hudson et al. 2017). Additionally, there appears to be a latitudinal gradient in heterozygosity within Britain, with the Welsh population in the south having the highest heterozygosity, followed by the English populations, and then the Scottish populations in the north. Private allelic richness was three times higher in Llyn Tegid compared to the second richest populations of Haweswater in England and Lomond in Scotland. We speculate that the latitudinal gradient in heterozygosity could have been generated during the postglacial recolonization of Britain as glaciers retreated northward. Similar patterns have been observed for mammals (Pope et al. 2006; Edwards et al. 2012) and plants (Sutherland et al. 2010). Lower genetic diversity at the range edge of species distributions, as European whitefish is in Britain at the extreme western reaches of the distribution, is a known phenomenon (Eckert et al. 2008), and in Europe there is a documented inverse correlation between distance from glacial refugium and contemporary genetic variation (e.g. Schmitt and Seitz 2002; Ficetola et al. 2007; Schmitt 2007; Dufresnes et al. 2014). Many populations in the British Isles have shown lower genetic diversity and/or high genetic differentiation as a result of postglacial recolonization events, including amphibians (Zeisset and Beebee 2001; Rowe et al. 2006), mammals (Dool et al. 2013; Montgomery et al. 2014), and fish (Bracken et al. 2015). A study using mitochondrial fragments found the Scottish Loch Lomond population to be part of the northern clade of European whitefish (Bernatchez and Dodson 1994), which has been hypothesised to have persisted during the most recent glaciation in a refugium west of the Ural mountain ridge (Østbye et al. 2005a, b), thus possibly explaining the observed lower genetic variation in the British populations. Furthermore, many continental whitefish populations exist in connected river systems (e.g. Winkler et al. 2011) and in the Baltic sea, possibly allowing for gene flow and potentially maintaining higher genetic diversity. An additional factor which could be contributing to the lower genetic diversity in the British populations is the lack of stocking or fisheries related translocations that have instead characterized many mainland European populations of whitefish (Eckman et al. 2007; Säisä et al. 2008; Dierking et al. 2014).
Evidence of genetic structuring among geographic areas
Analysis of population structure and molecular variance revealed three major genetic groups, which correspond to the populations in Scotland, England, and Wales. Our population differentiation estimate between Lochs Lomond and Eck in Scotland agrees with previous results (our FST = 0.063 vs FST = 0.056 of Adams et al. [2016]) and is much lower compared to the global FST of 0.388. The PCA supported this result of weak genetic structure between these two populations, and the DAPC analyses, with or without prior information on lake of origin, could not accurately assign several individuals to one of these lakes. Whitefish from these two lochs are quite distinct from each other in their morphology and ecology (Brown and Scott 1994; Pomeroy 1991; Etheridge et al. 2012b), and are believed to have been separated for thousands of years based on landscape features (Maitland 1970). The weak FST divergence between these two populations compared to the global FST could indicate a more recent divergence of the Loch Lomond and Loch Eck populations, compared to the divergence among most other British populations, or possibly indicates past introgression and gene flow.
The Welsh population of Llyn Tegid was more closely related to the English populations but formed its own genetic group, as indicated by the PCA and DAPC, and possessed many unique alleles. Coupling the higher genetic diversity of the Llyn Tegid population with the plausible south to north colonization route of whitefish after the ice retreat, it is reasonable to suggest the Welsh population was the first to settle in Britain.
In England we observed inter-lacustrine genetic differentiation that is hierarchically structured. The population of Red Tarn was the most divergent of the English group, separating on the NJ tree as sister to the other populations. In the DAPC analyses, individuals from Red Tarn formed a group that was clearly separated from the other English populations, and was somewhat close to the Welsh population. It has been suggested that the Red Tarn European whitefish population originated from human introduction (Macpherson 1892), given the high elevation of the lake, and founder effects might explain the high differentiation with the remaining English populations (Beaumont et al. 1995). However, there is no evidence of this introduction (Maitland et al. 1990), nor did we observe the bottleneck-associated reduced genetic diversity (Frankham et al. 2002) in the Red Tarn population when comparing it with the other English populations. Therefore, our analyses suggest Red Tarn is a natural and distinct population.
The next most divergent population was Haweswater, which also possesses the highest genetic diversity in England, and it is surpassed only by Llyn Tegid. The whitefish population in Haweswater has experienced severe declines due to anthropogenic water level fluctuations (the lake is used as water reservoir) and predation by cormorants (Winfield et al. 2013). Fortunately, our results suggest that this demographic crash is not reflected in the genetic diversity of the population, which remains relatively high. This suggests recovery will be possible if appropriate measures are put in place to prevent further reductions in overall population size.
The Brotherswater and Ullswater populations had the lowest inter-lacustrine differentiation of any two British populations, and DAPC analysis without prior information assigned individuals from both lakes to a single genetic cluster. These two lakes are connected by a short (length ~ 4 km) river, which despite no contemporary reports of sightings of whitefish along its length (Beaumont et al. 1995), could represent an ancestral corridor of gene flow for these populations. However, further studies are needed to discern the demographic events causing this pattern.
Our analysis of nuclear data supports the finding of previous research conducted with mitochondrial RFLPs (Hartley, 1995), in that the Welsh population has the highest genetic diversity, and that the seven British populations of whitefish cluster by geographic region. However, contrary to Hartley (1995) and previous studies based on electrophoretic analysis (Ferguson 1974; Ferguson et al. 1978) we find the Welsh population to be more closely related to, but not nested within, the English populations, as had been recorded by allozyme analysis (Beaumont et al. 1995). Finally, while previous studies recovered very low inter-population variability (Hartley 1995) in British European whitefish, our novel findings on hierarchical population genetic structuring, which were facilitated by the use of microsatellite loci, indicate high inter-population variation.
It is worth pointing out that all the lakes that contain European whitefish in Britain drain to the west of Britain into the Irish sea, where the post-glacial Lough Hibernia was located, and which Maitland (1970) believed to be the source of the British populations of C. lavaretus. If this were the case, it would support a monophyletic relationship of the British whitefish populations, but further studies including other European populations are needed to validate this hypothesis.
Evolutionary units of British Coregonus lavaretus
Our genetic analyses recover high genetic divergence between Scottish, English, and Welsh populations of European whitefish. Although there was some evidence of similar geographic clustering in phenotype observed by Etheridge et al. (2012a) based on the morphological traits proposed by Kottelat and Freyhof (2007) to distinguish the whitefish populations into the species C. clupeoides, C. stigmaticus, and C. pennantii, there was very considerable phenotypic overlap between individuals from these groups. Therefore, we argue that there is still not enough evidence to support the re-classification of European whitefish populations into three distinct species as was proposed in Kottelat and Freyhof (2007).
First, our inter-lacustrine genetic differentiation estimates within geographic areas contrast with the morphological results of Etheridge et al. (2012a). Those authors found the highest inter-population differentiation to be between Lomond and Eck in Scotland, while we observed low genetic differentiation between these two populations. Furthermore, they find Brotherswater individuals more morphologically similar to Red Tarn and Haweswater within England, while we found the lowest genetic differentiation in the dataset to be between Brotherswater and Ullswater. Our and Etheridge et al. (2012a) findings indicate that the morphological traits used to classify the three Coregonus species are too variable, perhaps influenced by local adaptation and/or phenotypic plasticity, and therefore not suitable for taxonomic purposes in these populations.
Second, while the level of genetic differentiation between whitefish populations in Britain is high, especially between Scottish and English lakes, it is within the levels of intra-population genetic differentiation observed for other temperate freshwater fish species as measured with microsatellite loci (e.g. Hänfling et al. 2002; Campos et al. 2006; Harris and Taylor 2010; Gordeeva et al. 2010; Taylor et al. 2011).
Therefore, while at this stage we cannot support the distinction of British C. lavaretus populations into three separate species, we recommend the delimitation of management unit (MU) status for each population, as these populations are demographically independent units, show restricted gene flow, high genetic differentiation and structuring (Funk et al. 2012). Furthermore, we propose to separate the Scottish, English, and Welsh populations into separate ESUs sensu Fraser and Bernatchez (2001) given their separate evolutionary histories.
Data availability
Data and scripts will be made available on Glasgow Enlighten repository upon publication.
Code availability
Code will be made available on Glasgow Enlighten repository upon publication.
References
Adams CE, Bean CW, Dodd JA et al (2016) Inter and intra-population phenotypic and genotypic structuring in the European whitefish Coregonus lavaretus, a rare freshwater fish in Scotland. J Fish Biol 88:580–594. https://doi.org/10.1111/jfb.12855
Adams C, Lyle A, Dodd J et al (2014) Translocation as a conservation tool: case studies from rare freshwater fishes in Scotland. Glas Nat 26:17–24
April J, Hanner RH, Dion-Côté A-M, Bernatchez L (2013) Glacial cycles as an allopatric speciation pump in north-eastern American freshwater fishes. Mol Ecol 22:409–422. https://doi.org/10.1111/mec.12116
Beaumont AR, Bray J, Murphy JM, Winfield IJ (1995) Genetics of whitefish and vendace in England and Wales. J Fish Biol 46:880–890. https://doi.org/10.1111/j.1095-8649.1995.tb01611.x
Bernatchez L, Dodson JJ (1994) Phylogenetic relationships among Palearctic and Nearctic withefish (Coregonus sp.) populations as revealed by mitochondrial DNA variation. Can J Fish Aquat Sci 51:240–250. https://doi.org/10.1139/f94-310
Bickford D, Lohman DJ, Sodhi NS et al (2007) Cryptic species as a window on diversity and conservation. Trends Ecol Evol 22:148–155
Bracken FSA, Hoelzel AR, Hume JB, Lucas MC (2015) Contrasting population genetic structure among freshwater-resident and anadromous lampreys: the role of demographic history, differential dispersal and anthropogenic barriers to movement. Mol Ecol 24:1188–1204. https://doi.org/10.1111/mec.13112
Brodersen J, Seehausen O (2014) Why evolutionary biologists should get seriously involved in ecological monitoring and applied biodiversity assessment programs. Evol Appl 7:968–983. https://doi.org/10.1111/eva.12215
Brown EAR, Scott DBC (1994) Life histories of the powan, Coregonus lavaretus (L.) (Salmonidae, Coregoninae) of Loch Lomond and Loch Eck. Hydrobiologia 290:121–133. https://doi.org/10.1007/BF00008959
Campos JL, Posada D, Morán P (2006) Genetic variation at MHC, mitochondrial and microsatellite loci in isolated populations of Brown trout (Salmo trutta). Conserv Genet 7:515–530. https://doi.org/10.1007/s10592-005-9063-z
Cavalli-Sforza LL, Edwards AW (1967) Phylogenetic analysis. Models and estimation procedures. Am J Hum Genet 19:233. https://doi.org/10.2307/2406616
de Oliveira LR, Hoffman JI, Hingst-Zaher E et al (2008) Morphological and genetic evidence for two evolutionarily significant units (ESUs) in the South American fur seal, Arctocephalus gazella. Conserv Genet 9:1451–1466. https://doi.org/10.1007/s10592-007-9473-1
Dierking J, Phelps L, Praebel K et al (2014) Anthropogenic hybridization between endangered migratory and commercially harvested stationary whitefish taxa (Coregonus spp.). Evol Appl 7:1068–1083. https://doi.org/10.1111/eva.12166
Dool SE, Puechmaille SJ, Dietz C et al (2013) Phylogeography and postglacial recolonization of Europe by Rhinolophus hipposideros: evidence from multiple genetic markers. Mol Ecol 22:4055–4070. https://doi.org/10.1111/mec.12373
Dufresnes C, Bertholet Y, Wassef J et al (2014) Sex-chromosome differentiation parallels postglacial range expansion in European tree frogs (Hyla arborea). Evolution 68:3445–3456. https://doi.org/10.1111/evo.12525
Eckert CG, Samis KE, Lougheed SC (2008) Genetic variation across species’ geographical ranges: the central-marginal hypothesis and beyond. Mol Ecol 17:1170–1188
Eckmann R, Kugler M, Ruhlé C (2007) Evaluating the success of large-scale whitefish stocking at Lake Constance. Adv Limnol 60:361–368
Edwards CJ, Soulsbury CD, Statham MJ et al (2012) Temporal genetic variation of the red fox, Vulpes vulpes, across western Europe and the British Isles. Quat Sci Rev 57:95–104. https://doi.org/10.1016/J.QUASCIREV.2012.10.010
Etheridge EC, Adams CE, Bean CW et al (2012a) Are phenotypic traits useful for differentiating among a priori Coregonus taxa? J Fish Biol 80:387–407. https://doi.org/10.1111/j.1095-8649.2011.03189.x
Etheridge EC, Bean CW, Maitland PS et al (2012b) Discontinuous infraspecific variation in ecological and morphological traits has consequences for conservation of powan (Coregonus lavaretus) in Scotland. Adv Limnol 505–517: https://doi.org/10.1127/ADVLIM/63/2012/505
Ferguson A (1974) The genetic relationships of the coregonid fishes of Britain and Ireland indicated by electrophoretic analysis of tissue proteins. J Fish Biol 6:311–315. https://doi.org/10.1111/j.1095-8649.1974.tb04548.x
Ferguson A, Himberg K-JM, Svardson G (1978) Systematics of the Irish pollan (Coregonus pollan Thompson): an electrophoretic comparison with other Holarctic Coregoninae. J Fish Biol 12:221–233. https://doi.org/10.1111/j.1095-8649.1978.tb04168.x
Ficetola GF, Garner TWJ, De Bernardi F (2007) Genetic diversity, but not hatching success, is jointly affected by postglacial colonization and isolation in the threatened frog, Rana latastei. Mol Ecol 16:1787–1797. https://doi.org/10.1111/j.1365-294X.2006.03198.x
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to conservation genetics. Cambridge University Press, Cambridge
Funk WC, McKay JK, Hohenlohe PA, Allendorf FW (2012) Harnessing genomics for delineating conservation units. Trends Ecol Evol 27:489–496. https://doi.org/10.1016/j.tree.2012.05.012
Gordeeva NV, Chukova EI, Oleinik AG (2010) Microsatellite genetic variation of Asian populations of Dolly Varden char. Hydrobiologia 650:133–144. https://doi.org/10.1007/s10750-010-0104-3
Goudet J (2005) HIERFSTAT, a package for R to compute and test hierarchical F-statistics. Mol Ecol Notes 5:184–186. https://doi.org/10.1111/j.1471-8286.2004.00828.x
Häkli K, Østbye K, Kahilainen KK et al (2018) Diversifying selection drives parallel evolution of gill raker number and body size along the speciation continuum of European whitefish. Ecol Evol 8:2617–2631. https://doi.org/10.1002/ece3.3876
Hänfling B, Hellemans B, Volckaert FAM, Carvalho GR (2002) Late glacial history of the cold-adapted freshwater fish Cottus gobio, revealed by microsatellites. Mol Ecol 11:1717–1729. https://doi.org/10.1046/j.1365-294X.2002.01563.x
Hansen MM, Fraser DJ, Als TD, Mensberg K-LD (2008) Reproductive isolation, evolutionary distinctiveness and setting conservation priorities: the case of European lake whitefish and the endangered North Sea houting (Coregonus spp.). BMC Evol Biol 8:137. https://doi.org/10.1186/1471-2148-8-137
Harris LN, Taylor EB (2010) Pleistocene glaciations and contemporary genetic diversity in a Beringian fish, the broad whitefish, Coregonus nasus (Pallas): Inferences from microsatellite DNA variation. J Evol Biol 23:72–86. https://doi.org/10.1111/j.1420-9101.2009.01858.x
Hartley SE (1995) Mitochondrial DNA analysis distinguishes between British populations of the whitefish. J Fish Biol 47:145–155. https://doi.org/10.1111/j.1095-8649.1995.tb06050.x
Hudson AG, Vonlanthen P, Bezault E, Seehausen O (2013) Genomic signatures of relaxed disruptive selection associated with speciation reversal in whitefish. BMC Evol Biol 13:108. https://doi.org/10.1186/1471-2148-13-108
Hudson AG, Lundsgaard-Hansen B, Lucek K et al (2017) Managing cryptic biodiversity: Fine-scale intralacustrine speciation along a benthic gradient in Alpine whitefish (Coregonus spp.). Evol Appl 10:251–266. https://doi.org/10.1111/eva.12446
Jacobs A, Carruthers M, Eckmann R et al (2019) Rapid niche expansion by selection on functional genomic variation after ecosystem recovery. Nat Ecol Evol 3:77–86. https://doi.org/10.1038/s41559-018-0742-9
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94. https://doi.org/10.1186/1471-2156-11-94
Kahilainen KK, Siwertsson A, Gjelland K et al (2011) The role of gill raker number variability in adaptive radiation of coregonid fish. Evol Ecol 25:573–588. https://doi.org/10.1007/s10682-010-9411-4
Kalinowski ST (2005) hp-rare 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5:187–189. https://doi.org/10.1111/j.1471-8286.2004.00845.x
Kamvar ZN, Tabima JF, Grünwald NJ (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281. https://doi.org/10.7717/peerj.281
Kottelat M, Freyhof J (2007) Handbook of European freshwater fishes. Kottelat, Berlin
Lindsey CC (1981) Stocks are chameleons: plasticity in gill rakers of coregonid fishes. Can J Fish Aquat Sci 38:1497–1506. https://doi.org/10.1139/f81-202
Mace GM (2004) The role of taxonomy in species conservation. Philos Trans R Soc London Ser B 359:711–719. https://doi.org/10.1098/rstb.2003.1454
Macpherson HA (1892) A vertebrate fauna of Lakeland : including Cumberland and Westmorland with Lancashire north of the sands. David Douglas
Maitland PS (1970) The origin and present distribution of Coregonus in the British Isles. In: Lindsay CC, Woods CS (eds) Biology of coregonid fishes. University of Manitoba Press, Winnipeg MB, pp 99–114
Maitland PS, Lyle AA (2013) Ex situ and in situ approaches, including assisted reproduction, for the conservation of native species of charr (Salmonidae) and whitefish (Coregonidae) in Scotland. Int Zoo Yearb 47:129–139. https://doi.org/10.1111/j.1748-1090.2012.00192.x
Maitland PS, Lyle AA, Barnett IKO (1990) Status of the schelly, Coregonus lavaretus (L.), in Red Tarn, Cumbria England. J Nat Hist 24:1067–1074. https://doi.org/10.1080/00222939000770641
Mayr G (2011) The phylogeny of charadriiform birds (shorebirds and allies)-reassessing the conflict between morphology and molecules. Zool J Linn Soc 161:916–934. https://doi.org/10.1111/j.1096-3642.2010.00654.x
Meirmans PG, Van Tienderenn PH (2004) genotype and genodive: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes 4:792–794. https://doi.org/10.1111/j.1471-8286.2004.00770.x
Moccetti P, Siwertsson A, Kjær R et al (2019) Contrasting patterns in trophic niche evolution of polymorphic Arctic charr populations in two subarctic Norwegian lakes. Hydrobiologia 840:281–299. https://doi.org/10.1007/s10750-019-3969-9
Mockford SW, Herman TB, Snyder M, Wright JM (2006) Conservation genetics of Blanding’s turtle and its application in the identification of evolutionarily significant units. Conserv Genet 8:209–219. https://doi.org/10.1007/s10592-006-9163-4
Montgomery WI, Provan J, McCabe AM, Yalden DW (2014) Origin of British and Irish mammals: disparate post-glacial colonisation and species introductions. Quat Sci Rev 98:144–165. https://doi.org/10.1016/J.QUASCIREV.2014.05.026
Østbye K, Bernatchez L, Næsje TF et al (2005a) Evolutionary history of the European whitefish Coregonus lavaretus (L.) species complex as inferred from mtDNA phylogeography and gill-raker numbers. Mol Ecol 14:4371–4387. https://doi.org/10.1111/j.1365-294X.2005.02737.x
Østbye K, Næsje TF, Bernatchez L et al (2005b) Morphological divergence and origin of sympatric populations of European whitefish (Coregonus lavaretus L.) in Lake Femund Norway. J Evol Biol 18:683–702. https://doi.org/10.1111/j.1420-9101.2004.00844.x
Ozerov MY, Himberg M, Aykanat T et al (2015) Generation of a neutral FST baseline for testing local adaptation on gill raker number within and between European whitefish ecotypes in the Baltic Sea basin. J Evol Biol 28:1170–1183. https://doi.org/10.1111/jeb.12645
Padial JM, Miralles A, De la Riva I, Vences M (2010) The integrative future of taxonomy. Front Zool 7:16. https://doi.org/10.1186/1742-9994-7-16
Patton JC, Gallaway BJ, Fechhelm RG, Cronin MA (1997) Genetic variation of microsatellite and mitochondrial DNA markers in broad whitefish (Coregonus nasus) in the Colville and Sagavanirktok rivers in northern Alaska. Can J Fish Aquat Sci 54:1548–1556. https://doi.org/10.1139/f97-062
Pomeroy PP (1991) A comparative assessment of temporal variation in diet of powan, Coregonus lavaretus (L.), from Loch Lomond and Loch Eck, Scotland, U.K. J Fish Biol 38:457–478. https://doi.org/10.1111/j.1095-8649.1991.tb03133.x
Pope LC, Domingo-Roura X, Erven K, Burke T (2006) Isolation by distance and gene flow in the Eurasian badger (Meles meles) at both a local and broad scale. Mol Ecol 15:371–386. https://doi.org/10.1111/j.1365-294X.2005.02815.x
Praebel K, Bean C, Dodd J et al (2019) Allelic losses and gains during translocations of a high conservation value fish, Coregonus lavaretus. SSRN Electron J. https://doi.org/10.2139/ssrn.3358962
Præbel K, Knudsen R, Siwertsson A et al (2013) Ecological speciation in postglacial European whitefish: rapid adaptive radiations into the littoral, pelagic, and profundal lake habitats. Ecol Evol 3:4970–4986. https://doi.org/10.1002/ece3.867
Praebel K, Westgaard J-I, Amundsen P-A et al (2013) A diagnostic tool for efficient analysis of the population structure, hybridization and conservation status of European whitefish (Coregonus lavaretus (L.)) and vendace (C. albula (L.)). Adv Limnol 64:247–255. https://doi.org/10.1127/1612-166X/2013/0064-0026
Quintela M, Berlin S, Wang B, Höglund J (2010) Genetic diversity and differentiation among Lagopus lagopus populations in Scandinavia and Scotland: evolutionary significant units confirmed by SNP markers. Mol Ecol 19:2380–2393. https://doi.org/10.1111/j.1365-294X.2010.04648.x
R Core Team (2019) The R project for statistical computing. In: Http://www.R-Project.Org/. https://www.r-project.org/. Accessed 18 Jun 2019
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225. https://doi.org/10.2307/2409177
Rogers SM, Marchand MH, Bernatchez L (2004) Isolation, characterization and cross-salmonid amplification of 31 microsatellite loci in the lake whitefish (Coregonus clupeaformis, Mitchill). Mol Ecol Notes 4:89–92. https://doi.org/10.1046/j.1471-8286.2003.00578.x
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8:103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Rowe G, Harris DJ, Beebee TJC (2006) Lusitania revisited: a phylogeographic analysis of the natterjack toad Bufo calamita across its entire biogeographical range. Mol Phylogenet Evol 39:335–346. https://doi.org/10.1016/J.YMPEV.2005.08.021
Ryder OA (1986) Species conservation and systematics: the dilemma of subspecies. Trends Ecol Evol 1:9–10. https://doi.org/10.1016/0169-5347(86)90059-5
Säisä M, Rönn J, Aho T et al (2008) Genetic differentiation among European whitefish ecotypes based on microsatellite data. Hereditas 145:69–83. https://doi.org/10.1111/j.0018-0661.2008.02050.x
Schmitt T (2007) Molecular biogeography of Europe: Pleistocene cycles and postglacial trends. Front Zool 4:11. https://doi.org/10.1186/1742-9994-4-11
Schmitt T, Seitz A (2002) Postglacial distribution area expansion of Polyommatus coridon (Lepidoptera: Lycaenidae) from its Ponto-Mediterranean glacial refugium. Heredity 89:20–26. https://doi.org/10.1038/sj.hdy.6800087
Siwertsson A, Knudsen R, Adams CE et al (2013) Parallel and non-parallel morphological divergence among foraging specialists in European whitefish (Coregonus lavaretus). Ecol Evol 3:1590–1602. https://doi.org/10.1002/ece3.562
Susnik S, Snoj A, Dovc P (1999) Microsatellites in grayling (Thymallus thymallus): Comparison of two geographically remote populations from the Danubian and Adriatic river basin in Slovenia. Mol Ecol 8:1756–1758
Sutherland BG, Belaj A, Nier S et al (2010) Molecular biodiversity and population structure in common ash (Fraxinus excelsior L.) in Britain: Implications for conservation. Mol Ecol 19:2196–2211. https://doi.org/10.1111/j.1365-294X.2009.04376.x
Svärdson G (1952) The coregonid problem. IV. The significance of scales and gillrakers report. Inst Freshw Res Drottningholm 33:204–232
Svärdson G (1979) Speciation of scandinavian coregonus. Inst Freshw Res 57:1–95
Taylor EB, Gow JL, Witt J, Zemlak R (2011) Connectivity among populations of pygmy whitefish (Prosopium coulterii) in northwestern North America inferred from microsatellite DNA analyses. Can J Zool 89:255–266. https://doi.org/10.1139/z10-114
Taylor EB (1999) Species pairs of north temperate freshwater fishes: Evolution, taxonomy, and conservation. Rev Fish Biol Fish 9:299–324. https://doi.org/10.1023/A:1008955229420
Thomas R, Fletcher JM, Ben JJ, Winfield IJ (2013) Assessment and conservation of gwyniad (Coregonus lavaretus (L.)) in Llyn Tegid, U.K.: persistence in the face of eutrophication, water level fluctuations and ruffe (Gymnocephalus cernuus (L.)) introduction. Adv Limnol 64:363–376. https://doi.org/10.1127/1612-166X/2013/0064-0016
Turgeon J, Estoup A, Bernatchez L (1999) Species flock in the North American Great Lakes: molecular ecology of Lake Nipigon ciscoes (Teleostei: Coregonidae: Coregonus). Evolution 53:1857–1871. https://doi.org/10.2307/2640446
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Vonlanthen P, Roy D, Hudson AG et al (2009) Divergence along a steep ecological gradient in lake whitefish (Coregonus sp.). J Evol Biol 22:498–514. https://doi.org/10.1111/j.1420-9101.2008.01670.x
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wennerström L, Laikre L, Ryman N et al (2013) Genetic biodiversity in the Baltic Sea: species-specific patterns challenge management. Biodivers Conserv 22:3045–3065. https://doi.org/10.1007/s10531-013-0570-9
Winfield IJ, Bean CW, Gorst J et al (2013) Assessment and conservation of whitefish (Coregonus lavaretus) in the U.K. Adv Limnol 64:305–321
Winkler KA, Weiss S (2008) Eighteen new tetranucleotide microsatellite DNA markers for Coregonus lavaretus cloned from an alpine lake population. Mol Ecol Resour 8:1055–1058. https://doi.org/10.1111/j.1755-0998.2008.02153.x
Winkler KA, Pamminger-Lahnsteiner B, Wanzenböck J, Weiss S (2011) Hybridization and restricted gene flow between native and introduced stocks of Alpine whitefish (Coregonus sp.) across multiple environments. Mol Ecol 20:456–472. https://doi.org/10.1111/j.1365-294X.2010.04961.x
Zeisset I, Beebee TJC (2001) Determination of biogeographical range: an application of molecular phylogeography to the European pool frog Rana lessonae. Proc R Soc Lond Ser B 268:933–938. https://doi.org/10.1098/rspb.2001.1600
Zhu Y, Wan Q-H, Yu B et al (2013) Patterns of genetic differentiation at MHC class I genes and microsatellites identify conservation units in the giant panda. BMC Evol Biol 13:227. https://doi.org/10.1186/1471-2148-13-227
Acknowledgements
We thank the Environment Agency, Natural Resources Wales, Scottish Natural Heritage and Natural England for providing licenses to sample the fish and funding the collection of material. We also thank the Norwegian Research Council (Grant No. 186320/V40) and UiT for funding.
Funding
Norwegian Research Council (Grant No. 186320/V40) and UiT.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
We declare no conflict of interest.
Ethical approval
License for fishing were provided by the Environment Agency, Natural Resources Wales, Scottish Natural Heritage and Natural England.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Crotti, M., Adams, C.E., Etheridge, E.C. et al. Geographic hierarchical population genetic structuring in British European whitefish (Coregonus lavaretus) and its implications for conservation. Conserv Genet 21, 927–939 (2020). https://doi.org/10.1007/s10592-020-01298-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-020-01298-y