
Abstract
Women with obesity who develop breast cancer have a worsened prognosis with diminished survival rates and increased rates of metastasis. Obesity is also associated with decreased breast cancer response to endocrine and chemotherapeutic treatments. Studies utilizing multiple in vivo models of obesity as well as human breast tumors have enhanced our understanding of how obesity alters the breast tumor microenvironment. Changes in the complement and function of adipocytes, adipose-derived stromal cells, immune cells, and endothelial cells and remodeling of the extracellular matrix all contribute to the rapid growth of breast tumors in the context of obesity. Interactions of these cells enhance secretion of cytokines and adipokines as well as local levels of estrogen within the breast tumor microenvironment that promote resistance to multiple therapies. In this review, we will discuss our current understanding of the impact of obesity on the breast tumor microenvironment, how obesity-induced changes in cellular interactions promote resistance to breast cancer treatments, and areas for development of treatment interventions for breast cancer patients with obesity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Obesity is a global epidemic, with the World Health Organization estimating that worldwide adult obesity has nearly tripled since 1975 [1, 2]. In the USA, 70% of adults have a body mass index (BMI) classified as overweight or obese [3]. Obesity significantly increases the risk for developing postmenopausal breast cancer [4,5,6]. On average, women undergo menopause between the ages of 45 and 55 years old, and approximately 80% of women diagnosed with breast cancer are over the age of 50, and the median age at diagnosis is 62 years of age [7, 8]. Postmenopausal women are most frequently diagnosed with breast cancers that express estrogen receptor alpha (ERα) and progesterone receptor (PR), and risk for diagnosis with this type of breast cancer is strongly correlated with increasing BMI [9,10,11,12]. Women with obesity are more likely to develop tumors of the luminal B molecular subtype, which are characterized by higher proliferation rates and reduced relapse-free survival [13, 14]. This subtype includes ERα+ and PR− tumors, in which 66% of patients fail to respond or develop early resistance to the selective ER modulator, tamoxifen [15, 16]. The association of obesity with other breast cancer subtypes in postmenopausal women is less clear. Epidemiological studies examining the impact of obesity on the incidence of triple-negative breast cancers (TNBCs), which lack expression of ERα, PR, and HER2, have shown conflicting results [11, 12, 17, 18].
In contrast, epidemiological studies suggest that obesity reduces breast cancer risk in the general population of women prior to menopause [5, 6, 12], suggesting a complex interaction between obesity and breast cancer risk. Similarly, while obesity increases the risk of ERα+ breast cancer in postmenopausal women, premenopausal women with obesity may have reduced risk for this breast cancer subtype [11, 19, 20]. However, the incidence of TNBC may be enhanced in younger women with obesity [17, 19,20,21]. Racial differences may also impact breast cancer risk in young women, as elevated BMI is correlated with increased breast cancer risk in premenopausal Asian women [22]. Obesity may also worsen the impact of other underlying risk factors for breast cancer including familial risk factors, leading to elevated breast cancer risk in selected groups of young women with obesity [17, 20, 21, 23, 24].
Regardless of age or menopausal status, breast cancer patients who are obese have a significantly worse overall and breast cancer-specific survival compared to patients with a BMI in the healthy range [9, 25,26,27]. At the time of diagnosis, breast cancer patients with obesity more frequently present with tumors of a higher grade, larger tumor size, and lymph node involvement compared to patients with a BMI in the healthy range [9, 25, 26, 28]. Long-term follow-up of breast cancer patients revealed that individuals who are obese also developed metastatic disease more rapidly after diagnosis with a higher frequency of distant recurrence than patients with a BMI in the healthy range (18.5–24.9 kg/m2) [25, 26, 28, 29]. Since obesity enhances progression, it has been hypothesized that increased tumor size and grade of the primary tumor at the time of diagnosis may contribute to the clinically observed increase in metastatic frequency [30, 31]. However, both clinical and pre-clinical studies have suggested that changes in the microenvironment of the tumor and surrounding breast tissue under conditions of obesity could promote expansion of metastasis-initiating cells, such as tumor cells that undergo a partial epithelial-to-mesenchymal transition (EMT), whereby tumor cells express mixed epithelial and mesenchymal genes [32,33,34,35,36,37]. Furthermore, systemic factors secreted from adipose tissue under conditions of obesity in the presence of a primary breast tumor could also enhance metastasis through recruitment of immune cell populations to distal metastatic sites [38,39,40,41]. Together, these studies suggest that obesity-induced changes in the tumor microenvironment may enhance tumor growth and aggressive characteristics leading to the clinically observed increased metastasis and worsened breast cancer survival in patients who are obese.
In addition to the increased risk of disease progression, breast cancer patients with BMI in the obese range are more likely to develop resistance to endocrine therapies and chemotherapies than women with BMI in the healthy range [25]. Postmenopausal women with a BMI greater than 30 kg/m2 and ERα+ breast cancers treated with aromatase inhibitors anastrozole or letrozole showed significantly decreased treatment efficacy compared to breast cancer patients with BMI in the healthy range [29, 42,43,44], yet no difference has been observed in the efficacy of the hormone therapy tamoxifen in breast cancer patients with differing BMI [43, 45]. While it has been hypothesized that the observed decrease in treatment efficacy of aromatase inhibitors in women with obesity could be due to an inadequate suppression of aromatase within adipose tissue [46], pre-clinical models have also suggested that alterations in the stroma within and surrounding breast tumors may also impact responsiveness [47, 48]. Obesity has also been associated with diminished efficacy of breast cancer chemotherapy [49,50,51,52], resulting in worse overall patient survival following treatment. Differences in response to chemotherapy may also be due in part to changes in the breast tumor microenvironment. Breast tumors from women with obesity have an elevated incidence of desmoplasia [53]. Desmoplastic tumors, which are characterized by increased numbers of cancer-associated fibroblasts (CAFs) within the stroma as well as deposition of fibrillar collagens, are associated with diminished survival in breast cancer patients [54,55,56,57]. Furthermore, desmoplasia in TNBC is associated with reduced relapse-free survival following chemotherapy [55, 56, 58, 59]. While other aspects of the breast tumor microenvironment have been recently reviewed [60], in this review, we will examine how obesity alters the cellular and extracellular structure of the tumor microenvironment to promote the growth of aggressive, treatment-resistant breast tumors.
2 Cancer-associated fibroblasts and adipocytes
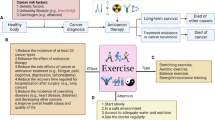
CAFs have diverse functions in the tumor microenvironment ranging from ECM deposition and remodeling to signaling interactions with cancer cells and surrounding immune cells. Although CAFs in breast tumors have been traditionally identified using the marker alpha-smooth muscle actin, CAFs are a heterogeneous population of cells that can be separated into subsets based on the expression of markers including fibroblast activation protein (FAP), fibroblast-specific protein-1 (FSP-1/S100A4), platelet-derived growth factor alpha (PDGFα) and PDGFβ [61,62,63,64]. CAFs have been suggested to originate from various sources, including resident fibroblasts, bone marrow-derived mesenchymal stem cells, pericytes, and tumor or endothelial cells that have undergone a mesenchymal transition [65, 66]. The impact of obesity on specific CAF populations within breast tumors is an emerging field. Obesity-induced changes in the function of stromal cells in the surrounding adipose tissue prior to tumor formation suggest that obesity could significantly alter the composition and function of CAF within the tumor stroma (Fig. 1).

Breast tumors from patients with obesity demonstrate increased tumor desmoplasia. Under conditions of obesity, breast tumors have more aligned collagen within and surrounding the tumor as well as increased ECM components, including hyaluronan. CAFs from multiple sources, including resident fibroblasts, adipocyte-derived fibroblasts, and adipose-derived stromal cells, are increased in the tumor microenvironment in obesity and promote aggressive cancer cell growth through secretion of growth factors, elevation of ECM remodeling enzymes, and local production of estrogen through aromatase activity. Adipocytes within the tumor microenvironment alter tumor cell metabolism by enhancing fatty acids, secretion of growth factors and adipokines, and further elevating local levels of estrogen. Together, these cells contribute to chemotherapy resistance and accelerated breast tumor growth observed in patients with obesity
Adipose tissue contains mesenchymal cells with the capacity for self-renewal and multipotent differentiation, including adipocytes, osteoblasts, and fibroblasts [67, 68]. Challenges exist in identifying and isolating adipose stem cells from other stromal cells present within adipose tissue, and studies examining adipose stem cells within the mammary gland frequently isolate a mixture of stromal cells, termed adipose-derived stromal cells. Adipose-derived stromal cells have the potential to differentiate into CAF within the tumor microenvironment, leading to mammary tumor progression [69], and obesity has been shown to promote adipose-derived stromal cell differentiation into myofibroblasts within tumors [53, 70, 71]. Adipose-derived stromal cells are expanded in obesity and can traffic from white adipose tissue into mammary tumors to promote growth [72]. Interestingly, obese breast cancer survivors have elevated circulating adipose-derived stromal cells compared to lean patients [73]. In pre-clinical models, increased numbers of these circulating cells play a role in distal metastases [72] through protection of circulating tumor cells from shear stress by enhancing cellular adhesions, preventing tumor cell apoptosis by activating cell survival pathways, and establishing metastatic niches at distal sites [74, 75].
Several co-culture experiments have demonstrated adipose-derived stromal cells from patients with obesity promote tumor cell migration, proliferation, and invasion [33, 34, 37, 76]. After exposure to adipose-derived stromal cells isolated from patients with obesity, breast cancer cells demonstrated increased ability to grow in suspension as tumorspheres, suggesting that the cancer cells had enhanced stem cell-like properties [34, 35, 37]. The increased invasive properties of tumor cells after exposure to adipose-derived stroma cells were abrogated by insulin-like growth factor-1 (IGF-1) neutralizing antibodies [37]. In a C(3)-Tag mouse model of basal breast cancer, smooth muscle actin-expressing myofibroblasts isolated from the mammary glands of obese mice produced elevated levels of hepatocyte growth factor (HGF), which significantly enhanced cancer cell proliferation and migration [70]. Adipose-derived stromal cells may also enhance the growth of ERα+ tumors. Adipose-derived stromal cells isolated from patients with obesity promoted secretion of leptin to regulate ERα+ MCF-7 tumor cell growth and aggressive tumor cell properties [33]. Silencing of leptin in adipose-derived stromal cells from patients with obesity led to reduced MCF-7 tumor cell expression of markers associated with EMT and reduced metastasis [33]. Additionally, adipose-derived stromal cells contribute to enhanced local levels of estrogen through expression of aromatase. Elevated levels of the inflammatory cytokine prostaglandin E2 in obesity led to downregulation of p53 in adipose-derived stromal cells resulting in increased aromatase expression and enhanced potential for ERα+ tumor formation [77]. Adipose-derived stromal cells may also contribute to treatment resistance within the breast tumor microenvironment. Adipose-derived stromal cells isolated from breast tissue from patients with obesity led to reduced sensitivity to the aromatase inhibitor, anastrozole, when co-cultured with MCF-7 cells in a 3D organotypic breast cancer model compared to adipose-derived stromal cells isolated from women with a healthy range BMI [47].
Breast adipose tissue exists adjacent to mammary ducts such that when breast tumors form, tumors are in close proximity to adipocytes. The proximity of cancer cells to adipocytes leads to the generation of fibroblast-like cells, termed adipocyte-derived fibroblasts, which may also be incorporated into the tumor stroma [78, 79]. Interestingly, adipocyte-derived fibroblasts do not express smooth muscle actin [78], which may contribute to the heterogeneity of markers observed in breast CAF. Loss of lipid droplets in adipocytes may be mediated in part through expression of stromelysin-3, which was found to be highly expressed in adipocytes at the invasive border of human breast tumors and diminished adipocyte differentiation in vitro [80]. It remains to be determined how obesity may alter the behavior of the adipocyte-derived fibroblasts within the tumor microenvironment.
Adipocytes have also been shown to enhance aggressive characteristics of tumor cells, which may contribute to tumor progression and metastasis. Co-culture of mature adipocytes with breast cancer cells promoted tumor cell proliferation [81,82,83,84], migration [81, 83, 85, 86], and invasion [83, 87]. When breast cancer cells were co-cultured with adipocytes isolated from patients with obesity, the capacity for tumor cell proliferation [32, 82], migration [32, 88], and invasion [88] was further increased. Numerous hypotheses have surfaced to delineate the mechanisms of how adipocytes isolated under obese conditions promote these phenotypes, including increased production of proinflammatory cytokines [87] and transcriptional regulation through microRNAs [83]. Balaban et al. demonstrated that breast cancer cells stimulated lipolysis of adipocytes isolated from obese patients in culture, leading cancer cells to accumulate adipocyte-derived fatty acids, which altered their cellular metabolism and induced proliferation and migration [32]. Obesity also regulates the expression of multiple adipokines through reduction of p16INK4A in adipocytes [88]. Exposure of breast tumor cells to secretions of obesity-activated adipocytes led to expression of EMT markers [88]. Promotion of breast cancer cell EMT has been associated with increased expression of forkhead box C2, twist-related protein-1, and N-cadherin, along with decreased expression of E-cadherin, in tumor cells [89]. Through changes in adipocytes induced by obesity, mature adipocytes may promote tumor cell motility and selection for tumor cells with more aggressive characteristics. Furthermore, adipocytes may play a critical role in promotion of the growth of ERα+ tumors through elevated aromatase expression, as differentiated adipocytes have fivefold higher aromatase activity than adipose-derived stromal cells [90].
Adipocytes may also contribute to diminished efficacy of endocrine and chemotherapy treatments. Crosstalk between breast cancer cells and adipocytes led to suppression of the anti-proliferative effect of tamoxifen through adipocyte-induced changes in cancer cell gene expression [84, 91]. Expression of fibroblast growth factor receptor-1 (FGFR-1) on tumor cells is a known mediator of endocrine treatment resistance and is associated with poor breast cancer patient outcomes [92]. Adipocyte-mediated expression of FGF-1, the ligand for FGFR-1, is elevated under conditions of obesity in mice and humans [92], suggesting that adipocytes may contribute to endocrine resistance through upregulation of FGF-1. In addition to endocrine therapy resistance, adipocytes from patients with obesity have been shown to promote doxorubicin chemotherapy resistance [93]. When co-culturing obese adipocytes with breast cancer cells, adipocytes promoted the production of cytoplasmic vesicles by cancer cells, which sequestered doxorubicin, and were then expelled in the extracellular space [93], resulting in chemotherapy resistance in the tumor cells. Together, these studies suggest that both CAF within tumors and surrounding adipocytes may contribute both to the growth of aggressive breast tumors as well as resistance to treatment with multiple therapeutic agents.
3 Extracellular matrix
The extracellular matrix (ECM) provides structural and mechanical support for breast tumors, influences the migration of tumor and immune cells through its physical properties [94, 95], and is composed of a complex meshwork of collagen fibers, glycoproteins, and proteoglycans [96]. Accumulating evidence suggests that the tumor ECM may promote resistance to chemotherapy, either by providing a protective barrier that reduces the concentration of anti-cancer drugs within tumors [97, 98] or by enhancing cancer cell survival [99]. Emerging evidence suggests that obesity significantly alters collagen alignment and ECM composition within breast tumors (Fig. 1). Collagen fibers, which are a major component of ECM, may impede tumor invasion by acting as a barrier against migration [100] or facilitate tumor cell invasion by acting as a directional scaffold for cellular movement based on the fiber orientation [101]. Tumor-associated collagen signatures (TACSs) of collagen fiber organization and alignment were identified in human breast tumors as a predictive marker for tumor progression and invasiveness [102]. TACS-3, which is a signature that reflects high local collagen density and perpendicular alignment of collagen fibers to the tumor boundary, has been shown to be an independent prognostic indicator for disease-free and disease-specific survival in breast cancer [103]. In breast tumors from women with obesity as well as mammary cell line-derived tumors in obese mice, collagen fibers were observed to be more aligned with each other within and surrounding the tumors compared to those of women with a healthy range BMI and lean mice [53, 104]. This collagen signature within breast tumors may also enhance metastasis, as breast tumor cells were observed migrating along aligned fibers and invading into the surrounding tissue [105]. Consistent with these observations, in a model utilizing transgenic mice with stabilized collagen fibers, ERα+ mammary tumors demonstrated significantly increased numbers of circulating tumor cells as well as increased metastasis [106]. Further studies are necessary to determine how targeting collagen alignment and organization could decrease metastasis in pre-clinical models of obesity-associated breast cancer.
In addition to collagen fiber alignment, mouse studies suggest that elevated stiffness in tumors contributes to cancer cell aggression and compromises treatment efficacy [107]. In vitro, ECM deposited by adipose-derived stromal cells isolated from the mammary glands of obese mice was stiffer than ECM deposited by adipose-derived stromal cells of lean mice [53]. Furthermore, differences in ECM production from adipose-derived stromal cells isolated from mammary tissue of obese mice promoted proliferation and migration of cultured preneoplastic mammary epithelial cells [53]. One possibility for differences in stiffness of the ECM may be due to increased expression of the collagen-crosslinking enzyme lysyl oxidase (LOX), which is enhanced in obese adipose tissue of both rats and humans [108, 109]. LOX overexpression in mouse mammary glands increased tissue stiffness, collagen deposition, and linearity of collagen fibers, while LOX inhibition resulted in less linear collagen fibers with decreased collagen deposition [107]. In a pre-clinical model of breast cancer, enhanced collagen expression and stabilization by LOX led to elevated tumor hypoxia, malignant signaling, and dysregulated angiogenesis [110]. In contrast, targeting LOX in tumors diminished chemotherapy resistance [110, 111]. Microdissected stromal cells from human breast tumors expressed the highest levels of LOX, which was significantly correlated with disease-specific mortality [112]. Together, these studies suggest that targeting of LOX may be a novel strategy to reduce ECM stiffness and potentially improve outcomes in breast cancer patients with obesity.
Obesity has also been shown to alter the composition of the ECM in multiple organs, including adipose tissue [113, 114]. Proteomics of the obese mammary gland and mammary tumors derived from multiple cancer cell lines identified a common signature of nine matrisome proteins, which were upregulated in both tumor and the obese mammary gland ECM, including collagen VI, collagen XII, fibronectin-1, laminin alpha subunit 5, vitronectin, tropoelastin, von Willebrand factor-1, galectin-1, and annexin A3 [115]. Increased deposition of collagen VI significantly enhanced the invasion of MDA-MB-231 and MDA-MB-468 TNBC cell lines [115]. Endotrophin, a cleavage product of collagen VI, promoted aggressive mammary tumors with high metastatic growth [116]. Within the ECM, hyaluronan is a linear polysaccharide that was considered to be an inert structural component; however, hyaluronan has been shown to activate kinase cascades in fibroblasts and bind to cell surface receptors such as CD44, which is expressed on the surface of aggressive breast cancer cells [117,118,119]. Hyaluronan expression was also observed to be significantly elevated in breast tissue from women with obesity and correlated with poor survival in breast cancer patients [120]. Another ECM component, heparanase, which is the sole mammalian endoglucuronidase that cleaves heparan sulfate in ECM, was identified as highly expressed in the ECM in obesity-associated human breast tumors [121]. Elevated heparanase may enhance local production of aromatase, the rate-limiting enzyme in estrogen biosynthesis through activation of inflammatory signaling in adipose tissue macrophages [121]. Changes in ECM composition surrounding breast tumors due to obesity could also impact the normal function of adipocytes and macrophages leading to further promotion of breast cancer growth [122, 123].
Obesity-induced changes in the ECM may also enhance tumor growth through release of growth factors that impact the tumor microenvironment [124]. In obese adipose tissue, transforming growth factor beta-1 (TGFβ1) concentrations are significantly enhanced [125,126,127], and TGFβ1 has been recognized as the most potent inducer of transformation of normal fibroblasts to CAF [128, 129]. TGFβ1 is produced as an inactive, latent form which complexes with latent TGFβ1 binding proteins as well as other matrix components including matricellular protein decorin within the ECM [130, 131]. In a high-fat diet model of obesity in mice, both decorin and TGFβ1 were significantly enhanced in the ECM surrounding mammary epithelial cells, and complexes of decorin and latent TGFβ1 were identified in ECM isolated from breast tissue from women with obesity [132]. Decorin is also significantly increased in the ECM of visceral and subcutaneous adipose tissue of obese patients [133]. Interestingly, loss of decorin within the ECM of ductal carcinoma in situ due to ECM remodeling is a marker for tumor progression and correlates with more aggressive disease [134, 135]. It is tempting to speculate that loss of decorin during tumor progression may increase TGFβ1 bioavailability in the tumor microenvironment. A number of growth factors, including IGF-1, FGF, and HGF, have also been found to associate with the ECM [136], and alterations in composition of the ECM due to obesity may alter the reservoir of growth factors stored in the ECM during tumor progression.
4 Immune cells
Obesity alters the compliment and function of multiple different immune cell types within adipose tissue, and many of these changes may also be reflected in the tumor microenvironment, impacting the response of the tumor to therapy (Fig. 2). Tumor-associated macrophages (TAMs) have long been accepted as potent tumor promoters, acting through a variety of mechanisms to enhance tumor cell proliferation and invasion [137, 138]. Multiple therapeutic strategies impacting TAM survival or function have moved to phase I and phase II clinical trials [139, 140]. While pre-clinical studies have shown promise for therapeutic efficacy, significant questions remain regarding identification of patients that are most likely to respond to these therapies. Obesity is associated with chronic, macrophage-driven inflammation in breast adipose tissue, and the presence of macrophages surrounding necrotic adipocytes to form crown-like structures is considered to be a hallmark of obesity [141,142,143]. Macrophages forming crown-like structures secrete inflammatory cytokines such as tumor necrosis factor alpha (TNFα), interleukin-1β (IL-1β), and IL-6 and express marker the CD11c [142, 143]. Macrophages have been classified into M1 and M2 categories based on expression of cytokines in response to ex vivo stimulation [144]. Recent transcriptomic and proteomic studies have suggested that there is a more complex range of macrophage activation states than those captured using the M1/M2 classification system [145]. Macrophages in mammary adipose tissue of obese mice also appear to have unique metabolic activation due to the uptake of fatty acids released by adipocytes, leading to a distinct proinflammatory phenotype [146,147,148]. These macrophages, termed metabolically activated macrophages, have been shown to enhance markers of cancer stem-like cells in TNBC cell lines and have been identified within the breasts of women with obesity [146]. However, single cell sequencing of adipose tissue macrophages in obesity has revealed multiple distinct macrophage subtypes [149], and it is currently unclear whether these different subtypes of macrophages are present and have divergent functions in the breast tumors of women with obesity.

Obesity enhances myeloid-lineage cells within breast tumors and metastases. Chronic macrophage-driven inflammation within obese adipose tissue enhances circulating numbers of myeloid-lineage cells. Within breast tumors under conditions of obesity, TAMs play multiple roles including enhanced secretion of tumor-promoting cytokines, collagen deposition and remodeling, and promotion of angiogenesis. MDSCs have immunosuppressive functions within the tumor microenvironment and are increased in obesity. Adipocytes further enhance angiogenesis within breast tumors through secretion of VEGF and multiple cytokines and growth factors. Within surrounding adipose tissue, leptin increases PD-1 expression within CD8 + T cells and promotes an exhaustion phenotype. Within lung tissue of obese patients, macrophages, neutrophils, and MDSC are increased, leading to elevated extravasation of tumor cells and increased metastatic disease
In mammary tumors of pre-clinical models, macrophages have been shown to promote cancer cells with cancer stem cell-like behavior, leading to increased metastasis and resistance to treatment with chemotherapy [150, 151]. Serum concentrations of TNFα are elevated in individuals with obesity [152], and macrophages are thought to be the major source of TNFα in obesity [153]. Treatment of TNBC cell lines with exogenous TNFα enhanced proliferation [154], which is hypothesized to contribute to rapid tumor growth in vivo. Depletion of macrophages using anti-F4/80 antibodies in obese tumor-bearing mice resulted in diminished tumor growth compared to tumors from obese mice treated with IgG antibodies [155]. Although tumor size was decreased in macrophage-depleted obese mice, cancer stem-like cells remained unchanged from those of control obese mice [155]. These data suggest that other factors altered by obesity may promote the accumulation of aggressive tumor cells, such as leptin secreted by adipocytes [35, 156]. Macrophages have also been associated with progression and early dissemination for metastasis in premalignant breast cancer lesions [157, 158]. However, depletion of macrophages early in tumor progression in obese mice in a p53-null model of mammary tumorigenesis led to increased DNA damage quantified in mammary epithelial cells [159], suggesting that macrophages may also serve numerous functions in the context of obesity. Further studies are necessary to determine how the function of TAM changes in the context of obesity in the tumor microenvironment.
Macrophages are directly influenced by the ECM, and changes in the ECM as a consequence of obesity could alter TAM function in the tumor microenvironment. When cultured on decellularized ECM isolated from adipose tissue of women with obesity, bone marrow-derived macrophages demonstrated a gene expression profile consistent with TAM expressing elevated transcripts of CD206 and arginase-1 compared to macrophages cultured on ECM from women with a healthy range BMI [123]. In the presence of IL-6 and collagen within the tumor microenvironment, a subset of FAP+ TAM expressed cytokines consistent with a wound healing response observed in macrophages isolated from wounded skin, and this gene expression profile was correlated with poor prognosis in breast cancer patients [160]. Given the elevated levels of collagen observed in breast tumors from women with obesity [53], it is possible that macrophages with this wound-healing signature could be similarly observed in breast tumors from women with obesity. In a pre-clinical model of breast cancer, the ECM component tenascin-C promoted an immunosuppressive TAM phenotype through toll-like receptor-4 (TLR4) signaling [161]. Expression of both tenascin-C and TLR4 was significantly increased in visceral fat of obese mice [162], which may suggest that these changes could be observed in breast adipose tissue surrounding tumors under conditions of obesity.
In addition to signaling changes in response to the ECM, macrophages may contribute to deposition of ECM in the tumor microenvironment under conditions of obesity. In obese adipose tissue, upregulation of the chemokine CCL2 led to chronic macrophage-driven inflammation [142, 163, 164]. Overexpression of CCL2 in the tumor microenvironment similarly led to increased TAM infiltration and collagen deposition [165, 166], which was abrogated by depletion of CD11b+ cells, including macrophages [166]. Furthermore, depletion of macrophages using clodronate-containing liposomes in mice bearing EO771 tumors resulted in altered collagen fibrillar microstructure as quantified using second harmonic generation imaging and immunofluorescence [167]. Together, these studies suggest that TAM can promote the organization and deposition of ECM within the tumor. In addition to exposure to tenascin-C altering the behavior of TAM to become more immunosuppressive [161], tenascin-C within the ECM also promoted the generation of TAM that exhibited increased synthesis and phosphorylation of collagen family members [168]. TAMs within tumors have also been observed to reorganize the collagen fibers favoring metastasis [167]. CCL5, another cytokine produced by adipocytes [169], has also been shown to enhance TAM recruitment into residual tumors in a conditional model of ErbB2 overexpression in mice, leading to increased collagen deposition within the tumor site and eventual recurrence of the tumor [170]. Together, these studies suggest that in obese patients, TAM may play a critical role in patients with obesity in promoting tumor recurrence and metastasis through remodeling the ECM in the tumor microenvironment.
Neutrophils have been shown to have both growth-promoting and growth inhibitory effects within the tumor microenvironment, which may be context-dependent [171]. In a retrospective study, a high circulating neutrophil-to-lymphocyte ratio in breast cancer patients with obesity was associated with a worse breast cancer-specific survival [172], which could reflect immunosuppressive functions. Within mammary tumors from mice fed a high-fat diet to induce obesity, neutrophil recruitment and tumor size were significantly increased compared to tumors from control mice [36], suggesting neutrophils may not only influence tumor initiation but also aid in progression under conditions of obesity. In culture, human breast cancer cell migration was stimulated by neutrophil secretions [173]. Although the role of neutrophils within the tumor microenvironment in obesity is not well understood, the role of neutrophils in the promotion of metastatic growth in obesity has been recently explored. Neutrophils have been implicated in obesity-associated lung metastasis by aiding in tumor cell seeding and growth through an IL-5 and granulocyte–macrophage-colony stimulating factor (GM-CSF)-dependent mechanism [38]. In a high-fat diet mouse model of obesity, neutralization of GM-CSF significantly decreased metastatic foci in lungs of obese tumor-bearing mice compared to lean mice [41]. This increased metastasis may occur in part through neutrophil-mediated impairment of vascular integrity under conditions of obesity, which enhanced cancer cell extravasation into the lung parenchyma [174]. Together, these results suggest that neutrophilia within the lungs induced by obesity plays a significant role in enhancing metastatic growth (Fig. 2).
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of myeloid progenitor cells whose normal maturation into macrophages, dendritic cells, and neutrophils is impaired in tumors [175, 176]. Due to a lack of specific markers to definitively separate neutrophils from some types of MDSC, questions remain about the distinctive function of this group separate from neutrophils [171]. MDSCs are rarely found in adipose tissue in homeostatic conditions [177], but increase under conditions of obesity [178,179,180]. Although MDSCs represent a low percentage in the total number of immune cells in tumors, these cells appear to play a vital role in immune surveillance. MDSCs act to suppress inflammation, inhibit CD8+ T cell proliferation, and promote CD8+ T cell apoptosis [181]. In obese mice bearing either EO771 or PY8119 mammary tumors, granulocytic MDSCs were significantly increased through a CXCR1-mediated pathway and promoted the apoptosis of CD8+ T cells both in culture and in vivo [182]. Depletion of MDSC in mammary tumors of high-fat diet-fed mice significantly increased CD8+ T cell recruitment into tumors and decreased tumor volume [180]. Together, these data suggest that MDSC may promote tumor growth during obesity by impairing CD8+ T cell response. Tumor-infiltrating MDSC also demonstrated increased fatty acid uptake and activated fatty acid oxidation [183], which enhanced the immunosuppressive characteristics of the MDSC [184]. Obesity may significantly increase both the number and function of MDSC within the tumor microenvironment, which has implications both for tumor progression as well as potential immunotherapy strategies.
T cells respond specifically to antigens expressed by tumor cells, leading to antitumor immunity. Obesity is associated with reduced dendritic cell function and antigen presentation in mice [185], suggesting that obesity may diminish the responsiveness of the adaptive immune system to antigens from cancer cells. CD8+ T cells are the primary cells that eliminate tumor cells, and high CD8+ T cell infiltration into TNBC is associated with good clinical outcomes in breast cancer patients [186, 187]. However, when BMI is considered a variable, increased lymphocyte infiltration into tumors is not predictive for better survival in breast cancer patients with obesity [188]. In another study, obese breast cancer patients demonstrated lower total T cell infiltration within tumors prior to treatment compared to women with a healthy range BMI [189]. Decreased T cell infiltration in mammary tumors has also been observed in pre-clinical models [155, 182, 190, 191], which may suggest reduced immunosurveillance in tumors under conditions of obesity. In a pre-clinical model, treatment of mammary tumor-bearing mice with 27-hydroxycholesterol resulted in fewer cytotoxic CD8+ T lymphocytes within the tumor microenvironment [192]. These results suggest that elevated circulating levels of cholesterol could play a role in diminished CD8+ T cell recruitment in obesity and are consistent with the observation that use of statins to decrease circulating cholesterol levels leads to diminished cancer mortality [193]. Obesity has also been associated with loss of T cell diversity and dysfunctional T cell responses to viruses and cancer [194, 195]. One dysfunctional T cell state is exhaustion, which is characterized by distinct epigenetic and transcriptional phenotypes, loss of effector functions, and prolonged and increased expression of inhibitory markers, such as programmed cell death protein-1 (PD-1). T cell exhaustion is thought to occur due to chronic antigen stimulation [196, 197]. While CD8+ effector T cells are recruited to adipose tissue during the early stages of obesity [198, 199], recent studies have suggested that these cells also express PD-1 and have diminished responses in the absence of tumors in obese mice [200]. These studies suggest that CD8+ T cell dysfunction may occur prior to tumor formation in obesity. Within tumors of obese mice, excess production of leptin led to an increase in fatty acid oxidation in CD8+ T cells and reduced interferon-gamma expression [201], which is an indicator of T cell exhaustion [202]. This data is supported by a study from Kado et al. which identified that obesity promoted a switch from non-exhausted PD-1 negative CD8+ T cells to exhausted PD-1+ CD8+ T cells in mammary tumors from obese mice [203]. In addition, the exhausted PD-1+ CD8+ T cells demonstrated increased expression of osteopontin [203], which has previously been implicated in tumor progression by regulating multiple pathways [204]. Together, these studies suggest that obesity diminishes both the number of CD8+ T cells within the tumor microenvironment and inhibits their function.
In addition to CD8+ T cells, obesity has been shown to impact other T cell subtypes which may promote breast cancer growth. Within obese adipose tissue, increased expression of TNFα has been shown to reduce CD4+ T cell function [205], suggesting that obesity may also inhibit the function of CD4+ T cells early during breast tumorigenesis. Natural killer (NK) cells, which are a critical part of the innate immune system, are impaired in patients with obesity through elevated circulating levels of leptin [206,207,208]. A prospective study demonstrated that dysfunctional NK cells were associated with an increased breast cancer incidence [209]. Furthermore, lipid accumulation in NK cells isolated from individuals with obesity was associated with the loss of NK cell cytotoxicity against tumor cells which could be restored through metabolic reprogramming [210], suggesting that changes in lipid metabolism associated with obesity may also alter the function of NK cells. Another T cell subtype which has innate immune cell function is γδ+ T cells. IL-17-secreting γδ+ T cells have been shown to promote primary tumor growth and metastasis, both in mice and in humans [211]. Pro-tumoral IL-17+ γδ+ T cells selectively showed high lipid uptake and intracellular lipid storage and were expanded in mammary tissue and tumors of obese mice [212]. Together, these studies suggest that obesity-induced changes in multiple immune cell populations enhance tumor growth and progression.
5 Endothelium
Expansion of adipose tissue is coordinated with its vascularization, and adipocytes secrete multiple pro-angiogenic factors including vascular endothelial growth factor (VEGF), FGF-2, IL-6, CCL2, and leptin to coordinate this process [213]. Increased levels of these pro-angiogenic factors are found in circulation in women, rats and mice associated with obesity [214,215,216], which may cooperatively enhance angiogenesis within breast tumors. Increased microvascular density within breast cancer is correlated with progression, metastasis, and reduced overall survival in breast cancer patients [217, 218]. Elevated microvascular density has also been observed in obesity and associated with rapid tumor growth in canine mammary cancer [219] as well as multiple pre-clinical models of obesity and breast cancer. In a mouse model of postmenopausal obesity, EO771 tumors in the mammary glands of obese ovariectomized mice demonstrated significantly increased blood vessel density mediated by elevated levels of VEGF secreted by surrounding adipocytes [220]. Similarly, MMTV-PyMT transgenic mice fed a high-fat diet developed tumors with increased microvascular density and recruitment of macrophages through elevated levels of CCL2 [221]. In an isocaloric diet model, mice fed a diet high in cholesterol demonstrated the highest growth rate of MDA-MB-231 tumors, which had the highest microvascular density [222], suggesting that elevated cholesterol levels could play a role in increased angiogenesis in obese individuals. In culture, leptin and IL-6 produced by obese adipocytes induced activation, proliferation, and migration of endothelial cells through upregulation of VEGF and VEGFR-2 [91, 223], suggesting that adipocytes secrete multiple factors that could enhance angiogenesis within the tumor microenvironment. While increased blood vessel density and angiogenesis may suggest that individuals with obesity could be candidates for anti-angiogenic therapeutics, breast cancer patients with obesity treated with anti-VEGF antibodies were less sensitive to anti-VEGF treatment due to increased systemic concentrations of IL-6 and FGF-2, which compensated for VEGF inhibition [224]. Similarly, treatment of tumor-bearing obese mice with anti-mouse VEGF antibodies demonstrated reduced sensitivity to treatment, which was determined to be due to elevated body weight rather than diet [224]. Together, these studies suggest that anti-angiogenesis therapies in obese breast cancer patients may need to target multiple pathways due to the upregulation of multiple angiogenic factors secreted by obese adipocytes.
Macrophages recruited to the obese tumor microenvironment may further stimulate tumor angiogenesis by providing soluble growth factors, matrix remodeling enzymes, and other bioactive molecules that promote the growth and migration of endothelial cells. Macrophages have been shown to secrete multiple pro-angiogenic factors including VEGF, TNFα, GM-CSF, and IL-6 [225]. Macrophages isolated from the mammary tumor microenvironment of obese mice demonstrated elevated tie-2 expression [226], which may suggest that obesity enriches for a subpopulation of macrophages that are known to promote angiogenesis. In a humanized mouse model, elevated CCL2 within the mammary tumor microenvironment enhanced macrophage recruitment, and depletion of macrophages led to decreased blood vessel density and diminished tumor growth [227]. Crosstalk between macrophages and adipocytes may also contribute to the increased angiogenesis observed in obese breast cancer. Co-culture of breast adipocytes with THP-1 macrophages stimulated expression of VEGF-A in macrophages [228]. In obese mice, IL-1β produced by macrophages enhanced expression of VEGF as well as angiopoietin-like 4 from adipocytes surrounding mammary tumors [229, 230]. Transplant of EO771 tumor cells into the mammary glands of angiopoietin-like 4-null mice resulted in significantly reduced tumor growth with limited blood vessel density [230]. Macrophages also express the receptor for leptin [231], and leptin secretion by adipocytes may enhance the angiogenic factors secreted by macrophages. Given the number of angiogenic factors that are produced by macrophages under conditions of obesity, therapeutically targeting macrophages rather than individual angiogenic factors may lead to improved anti-angiogenic responses.
6 Opportunities to improve therapeutic outcomes
The effects of weight loss on the risk for breast cancer recurrence are currently under investigation in long-term, clinical trials [232]; however, little is known about how weight loss may impact the tumor microenvironment and response to therapies. Mammary glands of obese mice that underwent weight loss demonstrated global epigenetic programming more similar to adipose tissue from obese mice than lean controls [233], suggesting that weight loss is insufficient to revert hypermethylation of genes in the mammary gland during the weight loss period examined. In adipose-derived stromal cells isolated from formerly obese mice, some changes in proliferation and viability were improved compared to those isolated from obese mice; however, impaired oxidative respiration was still observed [234]. Together, these data suggest that some obesity-induced changes in adipose-derived stromal cells could be reversed with weight loss, but not others, which could impact stromal cells that contribute to the formation of CAF. In obese mice, an 8-week weight loss period resulted in significantly decreased accumulation of macrophages in subcutaneous adipose tissue, suggesting that caloric restriction may resolve some inflammation associated with obesity [235]. Similarly, weight reduction significantly reduced the number of crown-like structures present within the mammary glands of formerly obese mice; however, expression of inflammatory molecules, including IL-6, TNF-α, and IL-1β, remained significantly elevated in the mammary glands of weight loss-induced mice relative to lean mice [236]. Elevated expression of inflammatory mediators within the mammary gland could still promote angiogenesis in the tumor microenvironment. More studies are necessary to understand how weight loss may impact the function of cell populations within the tumor microenvironment that contribute to tumor aggressiveness and therapeutic resistance. Weight loss leads to decreased recruitment of macrophages into obese adipose tissue concurrent with reduced circulating myeloid-lineage cells systemically. In a pre-clinical model, formerly obese mice demonstrated significantly reduced lung neutrophilia, resulting in reduced mammary cancer metastatic foci within the lungs compared to obese mice [38]. Additional studies could shed light on how weight loss impacts breast cancer metastases at other sites, including bone, brain, and liver.
In addition to weight loss, other therapeutic strategies targeting the tumor microenvironment may be beneficial for obese breast cancer patients. Metformin is a commonly utilized drug in the treatment of type 2 diabetes and has limited toxicity when used in combination with other breast cancer therapies [237, 238]. In mammary tumor-bearing obese mice, metformin treatment reduced obesity-associated tumor growth and was associated with a marked decrease in angiogenesis within the tumor microenvironment [229]. Similarly, treatment of obese/diabetic mice with metformin during EO771 tumor growth resulted in reduced tumor growth rates with less aligned collagen, decreased vascularity, reduced numbers of CD206+ TAM, and diminished pulmonary metastasis [239]. Furthermore, treatment of ovariectomized rats with metformin decreased the growth of ERα+ mammary tumors and diminished aromatase expression in CD68+ macrophages [240]. Together, these results suggest that metformin may have therapeutic effects on multiple cell types within the breast tumor microenvironment. Multiple ongoing clinical trials are currently being conducted to assess the efficacy of metformin as an adjuvant therapy for early stage, invasive, and metastatic breast cancer. In the recent MA32-randomized breast cancer trial, patients with breast cancer treated with metformin resulted in mild weight reduction and improvement in metabolic parameters compared to patients receiving placebo [241]. Further analysis of the trial is ongoing to assess breast cancer outcomes and potential impact on the tumor microenvironment.
Another potential target for therapy is the renin-angiotensin axis. Although most characterized for regulation of blood pressure, the renin-angiotensin system has also been recognized for its pathological role in obesity and breast cancer [242]. Angiotensin expression is enhanced in obese adipocytes and has been linked to elevated inflammation and metabolic disturbance [242]. In a mouse model of breast cancer, tumor desmoplasia was significantly reduced and chemotherapy outcome was improved after the administration of angiotensin receptor blockers conjugated to nanopolymers that degraded selectively in the tumor microenvironment [243]. Together, these results suggest that targeting this pathway may improve chemotherapeutic efficacy for women with obesity. Additionally, therapeutics targeting adipose-derived stromal cells are being developed in pre-clinical models. Novel killer peptides, which target an adipose-derived stromal cell-binding domain of decorin, demonstrate dose-dependent cytotoxic specificity for adipose-derived stromal cells [244, 245]. Su et al. demonstrated that when prostate cancer cells were xenografted into obese and lean mice, treatment with the killer peptide D-WAT significantly reduced prostate cancer cell invasion and tumor growth in obese mice compared to vehicle-treated obese and lean mice [246]. These results suggest that targeted reduction of adipose-derived stromal cells in obese mice reduces aggressive characteristics of tumors, which may improve response to clinical treatments.
Multiple strategies have also emerged to therapeutically target TAM. As CCL2 upregulation in obesity promotes macrophage recruitment into adipose tissue [142, 163, 164], inhibitors of CCL2 have been developed to reduce TAM within the tumor microenvironment and at sites of metastasis. In mice bearing late-stage MMTV-PyMT mammary tumors with spontaneous metastasis, treatment with CCL2 inhibitors reduced metastasis through diminished recruitment of metastasis-associated macrophages [247]. However, cessation of CCL2 therapy led to increased and rapid metastasis in multiple pre-clinical mammary tumor models through VEGF-mediated angiogenesis [248]. Elevated circulating levels of CCL2 following treatment could potentially be a significant problem in breast cancer patients with obesity. Colony-stimulating factor-1 (CSF-1) inhibitors have also been developed for clinical use and act to both diminish TAM recruitment as well as promote macrophage apoptosis [249]. The efficacy of this approach may be limited by CAF in the tumor microenvironment, which promoted an increase in MDSC within tumors in response to treatment with CSF-1R inhibitors [250]. Since MDSCs are increased under conditions of obesity [178,179,180], the efficacy of CSF-1R inhibitors should be examined in obese pre-clinical models. Other strategies to alter the function of macrophages may prove beneficial to breast cancer patients with obesity. In a model of pancreatic adenocarcinoma, PI3Kγ inhibition led to improved CD8+ T cell responses, diminished tumor cell metastasis, and reduced desmoplasia [251]. However, PI3Kγ has been shown to regulate lipid metabolism in adipose tissue macrophages, and downregulation of PI3K signaling may lead to elevated macrophage lipid burden and death and further contribute to metabolic pathogenesis in obesity [252].
Although obesity appears to have detrimental effects on the adaptive immune system, clinical studies have surprisingly revealed increased responses to immunotherapies in obese cancer patients. Immune checkpoint inhibitors, particularly agents targeting PD-1 or its ligand PD-L1, have been approved for treatment of various cancers [253]. For breast cancer patients, treatment of metastatic, TNBC with PD-1 inhibitors is efficacious in approximately 18% of patients [254]. The mechanisms underlying these limited treatment responses are not clear, and biomarkers identifying patients that may respond to these therapies have not been delineated. In a cohort of cancer patients treated with anti-PD-L1 therapy, a larger percentage of patients with a BMI greater than 30 kg/m2 had increased progression-free survival and overall survival compared to patients with a BMI less than 30 kg/m2 [255]. This trend for increased efficacy of immunotherapy observed in multiple types of solid tumors could also apply to TNBC patients with obesity [256], and recent studies have demonstrated improved immunotherapy responses in pre-clinical models of breast cancer in obesity [257]. The chronic inflammation observed in the mammary gland and tumor microenvironment as a consequence of obesity and resulting T cell exhaustion may lead to a microenvironment with greater responsiveness to treatment with checkpoint inhibitors. However, there is also evidence that patients with obesity have an increased incidence of immune-related adverse events when treated with PD-1 therapy that result in discontinuation of treatment [258]. Further clinical trials are necessary to determine how obesity impacts response to immunotherapy in breast cancer. Pre-clinical studies in this area will help guide new therapeutics for targeting the immune response to improve cancer outcomes for patients with obesity.
7 Conclusions
The tumor microenvironment is complex, with interactions among different cell types supporting the growth and malignancy of tumor cells. Obesity alters the function of multiple cell types within the tumor microenvironment, leading to the growth of breast tumors with increased propensity for therapy resistance and recurrence. While we are beginning to uncover how obesity shapes individual cell types within the tumor microenvironment, there is much to be discovered in how these cell types interact and are further modified by the hormonal changes of menopause. Furthermore, breast cancer is made up of multiple subtypes, each with distinct differences in the cell populations that make up the tumor microenvironment. Continued refinement of pre-clinical models to examine how obesity alters the function of stromal cells within different breast cancer subtypes is necessary to improve treatment responses for patients with obesity and identify potential targets for the development of new therapeutic interventions.
References
World Health Organization. Obesity and overweight. 2018. http://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight.
National Institute of Diabetes and Digestive and Kidney Diseases. Overweight & Obesity Statistics. 2017. https://www.niddk.nih.gov/health-information/health-statistics/overweight-obesity.
Mitchell, S., & Shaw, D. (2015). The worldwide epidemic of female obesity. Best Practice & Research. Clinical Obstetrics & Gynaecology, 29(3), 289–299. https://doi.org/10.1016/j.bpobgyn.2014.10.002
Lauby-Secretan, B., Scoccianti, C., Loomis, D., Grosse, Y., Bianchini, F., & Straif, K. (2016). Body fatness and cancer–Viewpoint of the IARC Working Group. New England Journal of Medicine, 375(8), 794–798. https://doi.org/10.1056/NEJMsr1606602
van den Brandt, P. A., Spiegelman, D., Yaun, S. S., Adami, H. O., Beeson, L., Folsom, A. R., et al. (2000). Pooled analysis of prospective cohort studies on height, weight, and breast cancer risk. American Journal of Epidemiology, 152(6), 514–527. https://doi.org/10.1093/aje/152.6.514
Lahmann, P. H., Hoffmann, K., Allen, N., van Gils, C. H., Khaw, K. T., Tehard, B., et al. (2004). Body size and breast cancer risk: Findings from the European Prospective Investigation into Cancer And Nutrition (EPIC). International Journal of Cancer, 111(5), 762–771. https://doi.org/10.1002/ijc.20315
DeSantis, C. E., Ma, J., Gaudet, M. M., Newman, L. A., Miller, K. D., Goding Sauer, A., et al. (2019). Breast cancer statistics, 2019. CA: A Cancer Journal for Clinicians, 69(6), 438–451. https://doi.org/10.3322/caac.21583
DeSantis, C. E., Ma, J., Goding Sauer, A., Newman, L. A., & Jemal, A. (2017). Breast cancer statistics, 2017, racial disparity in mortality by state. CA: A Cancer Journal for Clinicians, 67(6), 439–448. https://doi.org/10.3322/caac.21412
Neuhouser, M. L., Aragaki, A. K., Prentice, R. L., Manson, J. E., Chlebowski, R., Carty, C. L., et al. (2015). Overweight, obesity, and postmenopausal invasive breast cancer risk: A secondary analysis of the Women’s Health Initiative Randomized Clinical Trials. JAMA Oncology, 1(5), 611–621. https://doi.org/10.1001/jamaoncol.2015.1546
Sebastiani, F., Cortesi, L., Sant, M., Lucarini, V., Cirilli, C., De Matteis, E., et al. (2016). Increased incidence of breast cancer in postmenopausal women with high body mass index at the Modena Screening Program. Journal of Breast Cancer, 19(3), 283–291. https://doi.org/10.4048/jbc.2016.19.3.283
Suzuki, R., Orsini, N., Saji, S., Key, T. J., & Wolk, A. (2009). Body weight and incidence of breast cancer defined by estrogen and progesterone receptor status–A meta-analysis. International Journal of Cancer, 124(3), 698–712. https://doi.org/10.1002/ijc.23943
White, A. J., Nichols, H. B., Bradshaw, P. T., & Sandler, D. P. (2015). Overall and central adiposity and breast cancer risk in the Sister Study. Cancer, 121(20), 3700–3708. https://doi.org/10.1002/cncr.29552
Parker, J. S., Mullins, M., Cheang, M. C., Leung, S., Voduc, D., Vickery, T., et al. (2009). Supervised risk predictor of breast cancer based on intrinsic subtypes. Journal of Clinical Oncology, 27(8), 1160–1167. https://doi.org/10.1200/JCO.2008.18.1370
Gaudet, M. M., Press, M. F., Haile, R. W., Lynch, C. F., Glaser, S. L., Schildkraut, J., et al. (2011). Risk factors by molecular subtypes of breast cancer across a population-based study of women 56 years or younger. Breast Cancer Research and Treatment, 130(2), 587–597. https://doi.org/10.1007/s10549-011-1616-x
Clarke, R., Liu, M. C., Bouker, K. B., Gu, Z., Lee, R. Y., Zhu, Y., et al. (2003). Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene, 22(47), 7316–7339. https://doi.org/10.1038/sj.onc.1206937
Osborne, C. K., & Schiff, R. (2003). Growth factor receptor cross-talk with estrogen receptor as a mechanism for tamoxifen resistance in breast cancer. Breast, 12(6), 362–367. https://doi.org/10.1016/s0960-9776(03)00137-1
Chen, L., Cook, L. S., Tang, M. T., Porter, P. L., Hill, D. A., Wiggins, C. L., et al. (2016). Body mass index and risk of luminal, HER2-overexpressing, and triple negative breast cancer. Breast Cancer Research and Treatment, 157(3), 545–554. https://doi.org/10.1007/s10549-016-3825-9
Ritte, R., Lukanova, A., Berrino, F., Dossus, L., Tjønneland, A., Olsen, A., et al. (2012). Adiposity, hormone replacement therapy use and breast cancer risk by age and hormone receptor status: A large prospective cohort study. Breast Cancer Research, 14(3), R76. https://doi.org/10.1186/bcr3186
Willett, W. C., Browne, M. L., Bain, C., Lipnick, R. J., Stampfer, M. J., Rosner, B., et al. (1985). Relative weight and risk of breast cancer among premenopausal women. American Journal of Epidemiology, 122(5), 731–740. https://doi.org/10.1093/oxfordjournals.aje.a114156
Sahin, S., Erdem, G. U., Karatas, F., Aytekin, A., Sever, A. R., Ozisik, Y., et al. (2017). The association between body mass index and immunohistochemical subtypes in breast cancer. Breast, 32, 227–236. https://doi.org/10.1016/j.breast.2016.09.019
Pierobon, M., & Frankenfeld, C. L. (2013). Obesity as a risk factor for triple-negative breast cancers: A systematic review and meta-analysis. Breast Cancer Research and Treatment, 137(1), 307–314. https://doi.org/10.1007/s10549-012-2339-3
Amadou, A., Hainaut, P., & Romieu, I. (2013). Role of obesity in the risk of breast cancer: Lessons from anthropometry. Journal of Oncology, 2013, 19. https://doi.org/10.1155/2013/906495
Harris, H. R., Willett, W. C., Terry, K. L., & Michels, K. B. (2011). Body fat distribution and risk of premenopausal breast cancer in the Nurses’ Health Study II. Journal of the National Cancer Institute, 103(3), 273–278. https://doi.org/10.1093/jnci/djq500
Cecchini, R. S., Costantino, J. P., Cauley, J. A., Cronin, W. M., Wickerham, D. L., Land, S. R., et al. (2012). Body mass index and the risk for developing invasive breast cancer among high-risk women in NSABP P-1 and STAR breast cancer prevention trials. Cancer Prevention Research (Philadelphia, Pa.), 5(4), 583–592. https://doi.org/10.1158/1940-6207.Capr-11-0482
Ewertz, M., Jensen, M. B., Gunnarsdottir, K. A., Hojris, I., Jakobsen, E. H., Nielsen, D., et al. (2011). Effect of obesity on prognosis after early-stage breast cancer. Journal of Clinical Oncology, 29(1), 25–31. https://doi.org/10.1200/JCO.2010.29.7614
Loi, S., Milne, R. L., Friedlander, M. L., McCredie, M. R., Giles, G. G., Hopper, J. L., et al. (2005). Obesity and outcomes in premenopausal and postmenopausal breast cancer. Cancer Epidemiology, Biomarkers & Prevention, 14(7), 1686–1691. https://doi.org/10.1158/1055-9965.epi-05-0042
Chan, D. S., Vieira, A. R., Aune, D., Bandera, E. V., Greenwood, D. C., McTiernan, A., et al. (2014). Body mass index and survival in women with breast cancer-systematic literature review and meta-analysis of 82 follow-up studies. Annals of Oncology, 25(10), 1901–1914. https://doi.org/10.1093/annonc/mdu042
Biglia, N., Peano, E., Sgandurra, P., Moggio, G., Pecchio, S., Maggiorotto, F., et al. (2013). Body mass index (BMI) and breast cancer: Impact on tumor histopathologic features, cancer subtypes and recurrence rate in pre and postmenopausal women. Gynecological Endocrinology, 29(3), 263–267. https://doi.org/10.3109/09513590.2012.736559
Sestak, I., Distler, W., Forbes, J. F., Dowsett, M., Howell, A., & Cuzick, J. (2010). Effect of body mass index on recurrences in tamoxifen and anastrozole treated women: An exploratory analysis from the ATAC trial. Journal of Clinical Oncology, 28(21), 3411–3415. https://doi.org/10.1200/jco.2009.27.2021
Koscielny, S., Tubiana, M., Lê, M. G., Valleron, A. J., Mouriesse, H., Contesso, G., et al. (1984). Breast cancer: Relationship between the size of the primary tumour and the probability of metastatic dissemination. British Journal of Cancer, 49(6), 709–715. https://doi.org/10.1038/bjc.1984.112
Minn, A. J., Gupta, G. P., Padua, D., Bos, P., Nguyen, D. X., Nuyten, D., et al. (2007). Lung metastasis genes couple breast tumor size and metastatic spread. Proceedings of the National Academy of Sciences of the United States of America, 104(16), 6740–6745. https://doi.org/10.1073/pnas.0701138104
Balaban, S., Shearer, R. F., Lee, L. S., van Geldermalsen, M., Schreuder, M., Shtein, H. C., et al. (2017). Adipocyte lipolysis links obesity to breast cancer growth: Adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab., 5, 1. https://doi.org/10.1186/s40170-016-0163-7
Strong, A. L., Ohlstein, J. F., Biagas, B. A., Rhodes, L. V., Pei, D. T., Tucker, H. A., et al. (2015). Leptin produced by obese adipose stromal/stem cells enhances proliferation and metastasis of estrogen receptor positive breast cancers. Breast Cancer Research, 17, 112. https://doi.org/10.1186/s13058-015-0622-z
Goto, H., Shimono, Y., Funakoshi, Y., Imamura, Y., Toyoda, M., Kiyota, N., et al. (2019). Adipose-derived stem cells enhance human breast cancer growth and cancer stem cell-like properties through adipsin. Oncogene, 38(6), 767–779. https://doi.org/10.1038/s41388-018-0477-8
Picon-Ruiz, M., Pan, C., Drews-Elger, K., Jang, K., Besser, A. H., Zhao, D., et al. (2016). Interactions between adipocytes and breast cancer cells stimulate cytokine production and drive Src/Sox2/miR-302b-mediated malignant progression. Cancer Research, 76(2), 491–504. https://doi.org/10.1158/0008-5472.can-15-0927
Bousquenaud, M., Fico, F., Solinas, G., Rüegg, C., & Santamaria-Martínez, A. (2018). Obesity promotes the expansion of metastasis-initiating cells in breast cancer. Breast Cancer Research, 20(1), 104. https://doi.org/10.1186/s13058-018-1029-4
Hillers, L. E., D’Amato, J. V., Chamberlin, T., Paderta, G., & Arendt, L. M. (2018). Obesity-activated adipose-derived stromal cells promote breast cancer growth and invasion. Neoplasia, 20(11), 1161–1174. https://doi.org/10.1016/j.neo.2018.09.004
Quail, D. F., Olson, O. C., Bhardwaj, P., Walsh, L. A., Akkari, L., Quick, M. L., et al. (2017). Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nature Cell Biology, 19(8), 974–987. https://doi.org/10.1038/ncb3578
Hillers-Ziemer LE, Williams AE, Janquart A, Grogan C, Thompson V, Sanchez A et al. 2021 Obesity-activated lung stromal cells promote myeloid lineage cell accumulation and breast cancer metastasis. Cancers (Basel).;13(5). doi:https://doi.org/10.3390/cancers13051005.
Nagahashi, M., Yamada, A., Katsuta, E., Aoyagi, T., Huang, W. C., Terracina, K. P., et al. (2018). Targeting the SphK1/S1P/S1PR1 axis that links obesity, chronic inflammation, and breast cancer metastasis. Cancer Research, 78(7), 1713–1725. https://doi.org/10.1158/0008-5472.Can-17-1423
Reggiani, F., Labanca, V., Mancuso, P., Rabascio, C., Talarico, G., Orecchioni, S., et al. (2017). Adipose progenitor cell secretion of GM-CSF and MMP9 promotes a stromal and immunological microenvironment that supports breast cancer progression. Cancer Research, 77(18), 5169–5182. https://doi.org/10.1158/0008-5472.can-17-0914
Gnant, M., Pfeiler, G., Stöger, H., Mlineritsch, B., Fitzal, F., Balic, M., et al. (2013). The predictive impact of body mass index on the efficacy of extended adjuvant endocrine treatment with anastrozole in postmenopausal patients with breast cancer: An analysis of the randomised ABCSG-6a trial. British Journal of Cancer, 109(3), 589–596. https://doi.org/10.1038/bjc.2013.367
Pfeiler, G., Königsberg, R., Fesl, C., Mlineritsch, B., Stoeger, H., Singer, C. F., et al. (2011). Impact of body mass index on the efficacy of endocrine therapy in premenopausal patients with breast cancer: An analysis of the prospective ABCSG-12 trial. Journal of Clinical Oncology, 29(19), 2653–2659. https://doi.org/10.1200/jco.2010.33.2585
Wolters, R., Schwentner, L., Regierer, A., Wischnewsky, M., Kreienberg, R., & Wöckel, A. (2012). Endocrine therapy in obese patients with primary breast cancer: Another piece of evidence in an unfinished puzzle. Breast Cancer Research and Treatment, 131(3), 925–931. https://doi.org/10.1007/s10549-011-1874-7
Dignam, J. J., Wieand, K., Johnson, K. A., Fisher, B., Xu, L., & Mamounas, E. P. (2003). Obesity, tamoxifen use, and outcomes in women with estrogen receptor-positive early-stage breast cancer. Journal of the National Cancer Institute, 95(19), 1467–1476. https://doi.org/10.1093/jnci/djg060
Folkerd, E. J., Dixon, J. M., Renshaw, L., A’Hern, R. P., & Dowsett, M. (2012). Suppression of plasma estrogen levels by letrozole and anastrozole is related to body mass index in patients with breast cancer. Journal of Clinical Oncology, 30(24), 2977–2980. https://doi.org/10.1200/jco.2012.42.0273
Morgan, M. M., Livingston, M. K., Warrick, J. W., Stanek, E. M., Alarid, E. T., Beebe, D. J., et al. (2018). Mammary fibroblasts reduce apoptosis and speed estrogen-induced hyperplasia in an organotypic MCF7-derived duct model. Science and Reports, 8(1), 7139. https://doi.org/10.1038/s41598-018-25461-1
Schech, A., Yu, S., Goloubeva, O., McLenithan, J., & Sabnis, G. (2015). A nude mouse model of obesity to study the mechanisms of resistance to aromatase inhibitors. Endocrine-Related Cancer, 22(4), 645–656. https://doi.org/10.1530/erc-15-0168
Sparano, J. A., Wang, M., Zhao, F., Stearns, V., Martino, S., Ligibel, J. A., et al. (2012). Obesity at diagnosis is associated with inferior outcomes in hormone receptor-positive operable breast cancer. Cancer, 118(23), 5937–5946. https://doi.org/10.1002/cncr.27527
Karatas, F., Erdem, G. U., Sahin, S., Aytekin, A., Yuce, D., Sever, A. R., et al. (2017). Obesity is an independent prognostic factor of decreased pathological complete response to neoadjuvant chemotherapy in breast cancer patients. Breast, 32, 237–244. https://doi.org/10.1016/j.breast.2016.05.013
Litton, J. K., Gonzalez-Angulo, A. M., Warneke, C. L., Buzdar, A. U., Kau, S. W., Bondy, M., et al. (2008). Relationship between obesity and pathologic response to neoadjuvant chemotherapy among women with operable breast cancer. Journal of Clinical Oncology, 26(25), 4072–4077. https://doi.org/10.1200/jco.2007.14.4527
Iwase, T., Nakamura, R., Yamamoto, N., Yoshi, A., Itami, M., & Miyazaki, M. (2014). The effect of molecular subtype and body mass index on neo-adjuvant chemotherapy in breast cancer patients. Breast, 23(3), 264–272. https://doi.org/10.1016/j.breast.2013.11.008
Seo, B. R., Bhardwaj, P., Choi, S., Gonzalez, J., Andresen Eguiluz, R. C., Wang, K., et al. (2015). Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Science Translational Medicine, 7(301), 301ra130. https://doi.org/10.1126/scitranslmed.3010467
Beck, A. H., Sangoi, A. R., Leung, S., Marinelli, R. J., Nielsen, T. O., van de Vijver, M. J., et al. (2011). Systematic analysis of breast cancer morphology uncovers stromal features associated with survival. Sci Transl Med., 3(108), 108ra13. https://doi.org/10.1126/scitranslmed.3002564
de Kruijf, E. M., van Nes, J. G., van de Velde, C. J., Putter, H., Smit, V. T., Liefers, G. J., et al. (2011). Tumor-stroma ratio in the primary tumor is a prognostic factor in early breast cancer patients, especially in triple-negative carcinoma patients. Breast Cancer Research and Treatment, 125(3), 687–696. https://doi.org/10.1007/s10549-010-0855-6
Dekker, T. J., van de Velde, C. J., van Pelt, G. W., Kroep, J. R., Julien, J. P., Smit, V. T., et al. (2013). Prognostic significance of the tumor-stroma ratio: Validation study in node-negative premenopausal breast cancer patients from the EORTC perioperative chemotherapy (POP) trial (10854). Breast Cancer Research and Treatment, 139(2), 371–379. https://doi.org/10.1007/s10549-013-2571-5
Moorman, A. M., Vink, R., Heijmans, H. J., van der Palen, J., & Kouwenhoven, E. A. (2012). The prognostic value of tumour-stroma ratio in triple-negative breast cancer. European Journal of Surgical Oncology, 38(4), 307–313. https://doi.org/10.1016/j.ejso.2012.01.002
Dekker, T. J., Charehbili, A., Smit, V. T., ten Dijke, P., Kranenbarg, E. M., van de Velde, C. J., et al. (2015). Disorganised stroma determined on pre-treatment breast cancer biopsies is associated with poor response to neoadjuvant chemotherapy: Results from the NEOZOTAC trial. Molecular Oncology, 9(6), 1120–1128. https://doi.org/10.1016/j.molonc.2015.02.001
Hagenaars, S. C., de Groot, S., Cohen, D., Dekker, T. J. A., Charehbili, A., Meershoek-Klein Kranenbarg, E., et al. (2021). Tumor-stroma ratio is associated with Miller-Payne score and pathological response to neoadjuvant chemotherapy in HER2-negative early breast cancer. International Journal of Cancer, 149(5), 1181–1188. https://doi.org/10.1002/ijc.33700
Tumor Microenvironment in Breast Cancer Thematic Issue. (2021). American Journal of Pathology, 191(8), 1327–1487.
Augsten, M. (2014). Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Frontiers in Oncology, 4, 62. https://doi.org/10.3389/fonc.2014.00062
Cortez, E., Roswall, P., & Pietras, K. (2014). Functional subsets of mesenchymal cell types in the tumor microenvironment. Seminars in Cancer Biology, 25, 3–9. https://doi.org/10.1016/j.semcancer.2013.12.010
Sugimoto, H., Mundel, T. M., Kieran, M. W., & Kalluri, R. (2006). Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biology & Therapy, 5(12), 1640–1646. https://doi.org/10.4161/cbt.5.12.3354
Paulsson, J., & Micke, P. (2014). Prognostic relevance of cancer-associated fibroblasts in human cancer. Seminars in Cancer Biology, 25, 61–68. https://doi.org/10.1016/j.semcancer.2014.02.006
Sahai, E., Astsaturov, I., Cukierman, E., DeNardo, D. G., Egeblad, M., Evans, R. M., et al. (2020). A framework for advancing our understanding of cancer-associated fibroblasts. Nature Reviews Cancer, 20(3), 174–186. https://doi.org/10.1038/s41568-019-0238-1
Louault, K., Li, R. R., & DeClerck, Y. A. (2020). Cancer-associated fibroblasts: Understanding their heterogeneity. Cancers (Basel)., 12(11), 3108. https://doi.org/10.3390/cancers12113108
Zuk, P. A., Zhu, M., Mizuno, H., Huang, J., Futrell, J. W., Katz, A. J., et al. (2001). Multilineage cells from human adipose tissue: Implications for cell-based therapies. Tissue Engineering, 7(2), 211–228. https://doi.org/10.1089/107632701300062859
Zuk, P. A., Zhu, M., Ashjian, P., De Ugarte, D. A., Huang, J. I., Mizuno, H., et al. (2002). Human adipose tissue is a source of multipotent stem cells. Molecular Biology of the Cell, 13(12), 4279–4295. https://doi.org/10.1091/mbc.E02-02-0105
Strong, A. L., Pei, D. T., Hurst, C. G., Gimble, J. M., Burow, M. E., & Bunnell, B. A. (2017). Obesity enhances the conversion of adipose-derived stromal/stem cells into carcinoma-associated fibroblast leading to cancer cell proliferation and progression to an invasive phenotype. Stem Cells Int., 2017, 9216502. https://doi.org/10.1155/2017/9216502
Sundaram, S., Freemerman, A. J., Johnson, A. R., Milner, J. J., McNaughton, K. K., Galanko, J. A., et al. (2013). Role of HGF in obesity-associated tumorigenesis: C3(1)-TAg mice as a model for human basal-like breast cancer. Breast Cancer Research and Treatment, 142(3), 489–503. https://doi.org/10.1007/s10549-013-2741-5
Wolfson, B., Zhang, Y., Gernapudi, R., Duru, N., Yao, Y., Lo, P. K., et al. (2016). High-fat diet promotes mammary gland myofibroblast differentiation through miR-140 downregulation. Molecular and Cellular Biology, 37(4), e00461-e516. https://doi.org/10.1128/mcb.00461-16
Zhang, Y., Daquinag, A. C., Amaya-Manzanares, F., Sirin, O., Tseng, C., & Kolonin, M. G. (2012). Stromal progenitor cells from endogenous adipose tissue contribute to pericytes and adipocytes that populate the tumor microenvironment. Cancer Research, 72(20), 5198–5208. https://doi.org/10.1158/0008-5472.can-12-0294
Ghosh, S., Hughes, D., Parma, D. L., Ramirez, A., & Li, R. (2014). Association of obesity and circulating adipose stromal cells among breast cancer survivors. Molecular Biology Reports, 41(5), 2907–2916. https://doi.org/10.1007/s11033-014-3146-1
Hurtado, P., Martínez-Pena, I., & Piñeiro, R. (2020). Dangerous liaisons: Circulating tumor cells (CTCs) and cancer-associated fibroblasts (CAFs). Cancers (Basel)., 12(10), 2861. https://doi.org/10.3390/cancers12102861
Wang, Z., Liu, J., Huang, H., Ye, M., Li, X., Wu, R., et al. (2021). Metastasis-associated fibroblasts: An emerging target for metastatic cancer. Biomark Res., 9(1), 47. https://doi.org/10.1186/s40364-021-00305-9
Strong, A. L., Strong, T. A., Rhodes, L. V., Semon, J. A., Zhang, X., Shi, Z., et al. (2013). Obesity associated alterations in the biology of adipose stem cells mediate enhanced tumorigenesis by estrogen dependent pathways. Breast Cancer Research, 15(5), R102. https://doi.org/10.1186/bcr3569
Wang, X., Docanto, M. M., Sasano, H., Lo, C., Simpson, E. R., & Brown, K. A. (2015). Prostaglandin E2 inhibits p53 in human breast adipose stromal cells: A novel mechanism for the regulation of aromatase in obesity and breast cancer. Cancer Research, 75(4), 645–655. https://doi.org/10.1158/0008-5472.Can-14-2164
Bochet, L., Lehuede, C., Dauvillier, S., Wang, Y. Y., Dirat, B., Laurent, V., et al. (2013). Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Research, 73(18), 5657–5668. https://doi.org/10.1158/0008-5472.CAN-13-0530
Jotzu, C., Alt, E., Welte, G., Li, J., Hennessy, B. T., Devarajan, E., et al. (2011). Adipose tissue derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor derived factors. Cellular Oncology, 34(1), 55–67. https://doi.org/10.1007/s13402-011-0012-1
Andarawewa, K. L., Motrescu, E. R., Chenard, M. P., Gansmuller, A., Stoll, I., Tomasetto, C., et al. (2005). Stromelysin-3 is a potent negative regulator of adipogenesis participating to cancer cell-adipocyte interaction/crosstalk at the tumor invasive front. Cancer Research, 65(23), 10862–10871. https://doi.org/10.1158/0008-5472.CAN-05-1231
Rosendahl, A. H., Bergqvist, M., Lettiero, B., Kimbung, S., & Borgquist, S. (2018). Adipocytes and obesity-related conditions jointly promote breast cancer cell growth and motility: Associations with CAP1 for prognosis. Frontiers in Endocrinology, 9, 689. https://doi.org/10.3389/fendo.2018.00689
Bergqvist, M., Elebro, K., Borgquist, S., & Rosendahl, A. H. (2021). Adipocytes under obese-like conditions change cell cycle distribution and phosphorylation profiles of breast cancer cells: The adipokine receptor CAP1 matters. Frontiers in Oncology, 11, 628653. https://doi.org/10.3389/fonc.2021.628653
Rajarajan, D., Selvarajan, S., Charan Raja, M. R., Kar Mahapatra, S., & Kasiappan, R. (2019). Genome-wide analysis reveals miR-3184-5p and miR-181c-3p as a critical regulator for adipocytes-associated breast cancer. Journal of Cellular Physiology, 234(10), 17959–17974. https://doi.org/10.1002/jcp.28428
Delort, L., Bougaret, L., Cholet, J., Vermerie, M., Billard, H., Decombat, C., et al. (2019). Hormonal therapy resistance and breast cancer: Involvement of adipocytes and leptin. Nutrients, 11(12), 2839. https://doi.org/10.3390/nu11122839
Carter, J. C., & Church, F. C. (2012). Mature breast adipocytes promote breast cancer cell motility. Experimental and Molecular Pathology, 92(3), 312–317. https://doi.org/10.1016/j.yexmp.2012.03.005
Nickel, A., Blücher, C., Kadri, O. A., Schwagarus, N., Müller, S., Schaab, M., et al. (2018). Adipocytes induce distinct gene expression profiles in mammary tumor cells and enhance inflammatory signaling in invasive breast cancer cells. Science and Reports, 8(1), 9482. https://doi.org/10.1038/s41598-018-27210-w
Dirat, B., Bochet, L., Dabek, M., Daviaud, D., Dauvillier, S., Majed, B., et al. (2011). Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Research, 71(7), 2455–2465. https://doi.org/10.1158/0008-5472.CAN-10-3323
Al-Khalaf, H. H., Amir, M., Al-Mohanna, F., Tulbah, A., Al-Sayed, A., & Aboussekhra, A. (2017). Obesity and p16(INK4A) downregulation activate breast adipocytes and promote their protumorigenicity. Molecular and Cellular Biology, 37(17), e00101-e117. https://doi.org/10.1128/mcb.00101-17
Yao-Borengasser, A., Monzavi-Karbassi, B., Hedges, R. A., Rogers, L. J., Kadlubar, S. A., & Kieber-Emmons, T. (2015). Adipocyte hypoxia promotes epithelial-mesenchymal transition-related gene expression and estrogen receptor-negative phenotype in breast cancer cells. Oncology Reports, 33(6), 2689–2694. https://doi.org/10.3892/or.2015.3880
Bellemare, V., Laberge, P., Noël, S., Tchernof, A., & Luu-The, V. (2009). Differential estrogenic 17beta-hydroxysteroid dehydrogenase activity and type 12 17beta-hydroxysteroid dehydrogenase expression levels in preadipocytes and differentiated adipocytes. Journal of Steroid Biochemistry and Molecular Biology, 114(3–5), 129–134. https://doi.org/10.1016/j.jsbmb.2009.01.002
Bougaret, L., Delort, L., Billard, H., Lequeux, C., Goncalves-Mendes, N., Mojallal, A., et al. (2017). Supernatants of adipocytes from obese versus normal weight women and breast cancer cells: In vitro impact on angiogenesis. Journal of Cellular Physiology, 232(7), 1808–1816. https://doi.org/10.1002/jcp.25701
Wellberg, E. A., Kabos, P., Gillen, A. E., Jacobsen, B. M., Brechbuhl, H. M., Johnson, S. J., et al. (2018). FGFR1 underlies obesity-associated progression of estrogen receptor-positive breast cancer after estrogen deprivation. JCI insight., 3(14), e120594. https://doi.org/10.1172/jci.insight.120594
Lehuédé, C., Li, X., Dauvillier, S., Vaysse, C., Franchet, C., Clement, E., et al. (2019). Adipocytes promote breast cancer resistance to chemotherapy, a process amplified by obesity: Role of the major vault protein (MVP). Breast Cancer Research, 21(1), 7. https://doi.org/10.1186/s13058-018-1088-6
Frantz, C., Stewart, K. M., & Weaver, V. M. (2010). The extracellular matrix at a glance. Journal of Cell Science, 123(Pt 24), 4195–4200. https://doi.org/10.1242/jcs.023820
Charras, G., & Sahai, E. (2014). Physical influences of the extracellular environment on cell migration. Nature Reviews Molecular Cell Biology, 15(12), 813–824. https://doi.org/10.1038/nrm3897
Lochter, A., & Bissell, M. J. (1995). Involvement of extracellular matrix constituents in breast cancer. Seminars in Cancer Biology, 6(3), 165–173. https://doi.org/10.1006/scbi.1995.0017
Netti, P. A., Berk, D. A., Swartz, M. A., Grodzinsky, A. J., & Jain, R. K. (2000). Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Research, 60(9), 2497–2503.
Senthebane, D. A., Rowe, A., Thomford, N. E., Shipanga, H., Munro, D., Mazeedi, M., et al. (2017). The role of tumor microenvironment in chemoresistance: To survive, keep your enemies closer. International Journal of Molecular Sciences, 18(7), 1586. https://doi.org/10.3390/ijms18071586
Aoudjit, F., & Vuori, K. (2012). Integrin signaling in cancer cell survival and chemoresistance. Chemotherapy Research and Practice, 2012, 283181. https://doi.org/10.1155/2012/283181
Grossman, M., Ben-Chetrit, N., Zhuravlev, A., Afik, R., Bassat, E., Solomonov, I., et al. (2016). Tumor cell invasion can be blocked by modulators of collagen fibril alignment that control assembly of the extracellular matrix. Cancer Research, 76(14), 4249–4258. https://doi.org/10.1158/0008-5472.Can-15-2813
Azimzade, Y., Saberi, A. A., & Sahimi, M. (2019). Regulation of migration of chemotactic tumor cells by the spatial distribution of collagen fiber orientation. Physical Review E, 99(6–1), 062414. https://doi.org/10.1103/PhysRevE.99.062414
Esbona, K., Yi, Y., Saha, S., Yu, M., Van Doorn, R. R., Conklin, M. W., et al. (2018). The presence of cyclooxygenase 2, tumor-associated macrophages, and collagen alignment as prognostic markers for invasive breast carcinoma patients. American Journal of Pathology, 188(3), 559–573. https://doi.org/10.1016/j.ajpath.2017.10.025
Conklin, M. W., Eickhoff, J. C., Riching, K. M., Pehlke, C. A., Eliceiri, K. W., Provenzano, P. P., et al. (2011). Aligned collagen is a prognostic signature for survival in human breast carcinoma. American Journal of Pathology, 178(3), 1221–1232. https://doi.org/10.1016/j.ajpath.2010.11.076
Zhang, Y., Baloglu, F. K., Hillers-Ziemer, L. E., Liu, Z., Lyu, B., Arendt, L. M., et al. (2020). Factors associated with obesity alter matrix remodeling in breast cancer tissues. Journal of Biomedial Optics, 25(1), 1–14. https://doi.org/10.1117/1.Jbo.25.1.014513
Provenzano, P. P., Eliceiri, K. W., Campbell, J. M., Inman, D. R., White, J. G., & Keely, P. J. (2006). Collagen reorganization at the tumor-stromal interface facilitates local invasion. Bmc Medicine, 4(1), 38. https://doi.org/10.1186/1741-7015-4-38
Barcus, C. E., O’Leary, K. A., Brockman, J. L., Rugowski, D. E., Liu, Y., Garcia, N., et al. (2017). Elevated collagen-I augments tumor progressive signals, intravasation and metastasis of prolactin-induced estrogen receptor alpha positive mammary tumor cells. Breast Cancer Research, 19(1), 9. https://doi.org/10.1186/s13058-017-0801-1
Levental, K. R., Yu, H., Kass, L., Lakins, J. N., Egeblad, M., Erler, J. T., et al. (2009). Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell, 139(5), 891–906. https://doi.org/10.1016/j.cell.2009.10.027
Miana, M., Galan, M., Martinez-Martinez, E., Varona, S., Jurado-Lopez, R., Bausa-Miranda, B., et al. (2015). The lysyl oxidase inhibitor β-aminopropionitrile reduces body weight gain and improves the metabolic profile in diet-induced obesity in rats. Disease Models & Mechanisms, 8(6), 543–551. https://doi.org/10.1242/dmm.020107
Pastel, E., Price, E., Sjoholm, K., McCulloch, L. J., Rittig, N., Liversedge, N., et al. (2018). Lysyl oxidase and adipose tissue dysfunction. Metabolism, 78, 118–127. https://doi.org/10.1016/j.metabol.2017.10.002
Rossow, L., Veitl, S., Vorlová, S., Wax, J. K., Kuhn, A. E., Maltzahn, V., et al. (2018). LOX-catalyzed collagen stabilization is a proximal cause for intrinsic resistance to chemotherapy. Oncogene, 37(36), 4921–4940. https://doi.org/10.1038/s41388-018-0320-2
Saatci, O., Kaymak, A., Raza, U., Ersan, P. G., Akbulut, O., Banister, C. E., et al. (2020). Targeting lysyl oxidase (LOX) overcomes chemotherapy resistance in triple negative breast cancer. Nature Communications, 11(1), 2416. https://doi.org/10.1038/s41467-020-16199-4
Maller, O., Drain, A. P., Barrett, A. S., Borgquist, S., Ruffell, B., Zakharevich, I., et al. (2021). Tumour-associated macrophages drive stromal cell-dependent collagen crosslinking and stiffening to promote breast cancer aggression. Nature Materials, 20(4), 548–559. https://doi.org/10.1038/s41563-020-00849-5
Barreto, S. C., Hopkins, C. A., Bhowmick, M., & Ray, A. (2015). Extracellular matrix in obesity - Cancer interactions. Hormone Molecular Biology and Clinical Investigation, 22(2), 63–77. https://doi.org/10.1515/hmbci-2015-0001
Lin, Chun, T. H., & Kang, L. (2016). Adipose extracellular matrix remodelling in obesity and insulin resistance. Biochemical Pharmacology, 119, 8–16. https://doi.org/10.1016/j.bcp.2016.05.005
Wishart, A. L., Conner, S. J., Guarin, J. R., Fatherree, J. P., Peng, Y., McGinn, R. A., et al. (2020). Decellularized extracellular matrix scaffolds identify full-length collagen VI as a driver of breast cancer cell invasion in obesity and metastasis. Sci Adv, 6(43), 3175.
Park, J., & Scherer, P. E. (2012). Adipocyte-derived endotrophin promotes malignant tumor progression. The Journal of Clinical Investigation, 122(11), 4243–4256. https://doi.org/10.1172/jci63930
Tolg, C., Messam, B. J., McCarthy, J. B., Nelson, A. C., & Turley, E. A. (2021). Hyaluronan functions in wound repair that are captured to fuel breast cancer progression. Biomolecules, 11(11), 1551. https://doi.org/10.3390/biom11111551
Sironen, R. K., Tammi, M., Tammi, R., Auvinen, P. K., Anttila, M., & Kosma, V. M. (2011). Hyaluronan in human malignancies. Experimental Cell Research, 317(4), 383–391. https://doi.org/10.1016/j.yexcr.2010.11.017
Schwertfeger, K. L., Cowman, M. K., Telmer, P. G., Turley, E. A., & McCarthy, J. B. (2015). Hyaluronan, inflammation, and breast cancer progression. Frontiers in Immunology, 6, 236. https://doi.org/10.3389/fimmu.2015.00236
Tiainen, S., Masarwah, A., Oikari, S., Rilla, K., Hämäläinen, K., Sudah, M., et al. (2020). Tumor microenvironment and breast cancer survival: Combined effects of breast fat, M2 macrophages and hyaluronan create a dismal prognosis. Breast Cancer Research and Treatment, 179(3), 565–575. https://doi.org/10.1007/s10549-019-05491-7
Hermano, E., Goldberg, R., Rubinstein, A. M., Sonnenblick, A., Maly, B., Nahmias, D., et al. (2019). Heparanase accelerates obesity-associated breast cancer progression. Cancer Research, 79(20), 5342–5354. https://doi.org/10.1158/0008-5472.Can-18-4058
Pope, B. D., Warren, C. R., Parker, K. K., & Cowan, C. A. (2016). Microenvironmental control of adipocyte fate and function. Trends in Cell Biology, 26(10), 745–755. https://doi.org/10.1016/j.tcb.2016.05.005
Springer, N. L., Iyengar, N. M., Bareja, R., Verma, A., Jochelson, M. S., Giri, D. D., et al. (2019). Obesity-associated extracellular matrix remodeling promotes a macrophage phenotype similar to tumor-associated macrophages. American Journal of Pathology, 189(10), 2019–2035. https://doi.org/10.1016/j.ajpath.2019.06.005
Flaumenhaft, R., & Rifkin, D. B. (1992). The extracellular regulation of growth factor action. Molecular Biology of the Cell, 3(10), 1057–1065. https://doi.org/10.1091/mbc.3.10.1057
Samad, F., Yamamoto, K., Pandey, M., & Loskutoff, D. J. (1997). Elevated expression of transforming growth factor-beta in adipose tissue from obese mice. Molecular Medicine, 3(1), 37–48.
Alessi, M. C., Bastelica, D., Morange, P., Berthet, B., Leduc, I., Verdier, M., et al. (2000). Plasminogen activator inhibitor 1, transforming growth factor-beta1, and BMI are closely associated in human adipose tissue during morbid obesity. Diabetes, 49(8), 1374–1380. https://doi.org/10.2337/diabetes.49.8.1374
Fain, J. N., Tichansky, D. S., & Madan, A. K. (2005). Transforming growth factor beta1 release by human adipose tissue is enhanced in obesity. Metabolism, 54(11), 1546–1551. https://doi.org/10.1016/j.metabol.2005.05.024
Shi, X., Young, C. D., Zhou, H., & Wang, X. (2020). Transforming growth factor-β signaling in fibrotic diseases and cancer-associated fibroblasts. Biomolecules, 10(12), 1666. https://doi.org/10.3390/biom10121666
Vaughan, M. B., Howard, E. W., & Tomasek, J. J. (2000). Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Experimental Cell Research, 257(1), 180–189. https://doi.org/10.1006/excr.2000.4869
Moses, H., & Barcellos-Hoff, M. H. (2011). TGF-beta biology in mammary development and breast cancer. Cold Spring Harbor Perspectives in Biology, 3(1), a003277. https://doi.org/10.1101/cshperspect.a003277
Horiguchi, M., Ota, M., & Rifkin, D. B. (2012). Matrix control of transforming growth factor-β function. Journal of Biochemistry, 152(4), 321–329. https://doi.org/10.1093/jb/mvs089
Chamberlin, T., Thompson, V., Hillers-Ziemer, L. E., Walton, B. N., & Arendt, L. M. (2020). Obesity reduces mammary epithelial cell TGFβ1 activity through macrophage-mediated extracellular matrix remodeling. The FASEB Journal, 34(6), 8611–8624. https://doi.org/10.1096/fj.202000228RR
Bolton, K., Segal, D., McMillan, J., Jowett, J., Heilbronn, L., Abberton, K., et al. (2008). Decorin is a secreted protein associated with obesity and type 2 diabetes. International Journal of Obesity, 32(7), 1113–1121. https://doi.org/10.1038/ijo.2008.41
Oda, G., Sato, T., Ishikawa, T., Kawachi, H., Nakagawa, T., Kuwayama, T., et al. (2012). Significance of stromal decorin expression during the progression of breast cancer. Oncology Reports, 28(6), 2003–2008. https://doi.org/10.3892/or.2012.2040
Van Bockstal, M., Lambein, K., Gevaert, O., De Wever, O., Praet, M., Cocquyt, V., et al. (2013). Stromal architecture and periductal decorin are potential prognostic markers for ipsilateral locoregional recurrence in ductal carcinoma in situ of the breast. Histopathol., 63(4), 520–533. https://doi.org/10.1111/his.12188
Taipale, J., & Keski-Oja, J. (1997). Growth factors in the extracellular matrix. The Faseb Journal, 11(1), 51–59. https://doi.org/10.1096/fasebj.11.1.9034166
Quail, D. F., & Joyce, J. A. (2013). Microenvironmental regulation of tumor progression and metastasis. Nature Medicine, 19(11), 1423–1437. https://doi.org/10.1038/nm.3394
Williams, C. B., Yeh, E. S., & Soloff, A. C. (2016). Tumor-associated macrophages: Unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer., 2, 15025. https://doi.org/10.1038/npjbcancer.2015.25
Cassetta, L., & Pollard, J. W. (2018). Targeting macrophages: Therapeutic approaches in cancer. Nature Reviews. Drug Discovery, 17(12), 887–904. https://doi.org/10.1038/nrd.2018.169
DeNardo, D. G., & Ruffell, B. (2019). Macrophages as regulators of tumour immunity and immunotherapy. Nature Reviews Immunology, 19(6), 369–382. https://doi.org/10.1038/s41577-019-0127-6
Morris, P. G., Hudis, C. A., Giri, D., Morrow, M., Falcone, D. J., Zhou, X. K., et al. (2011). Inflammation and increased aromatase expression occur in the breast tissue of obese women with breast cancer. Cancer Prevention Research (Philadelphia, Pa.), 4(7), 1021–1029. https://doi.org/10.1158/1940-6207.CAPR-11-0110
Weisberg, S. P., McCann, D., Desai, M., Rosenbaum, M., Leibel, R. L., & Ferrante, A. W. (2003). Obesity is associated with macrophage accumulation in adipose tissue. The Journal of Clinical Investigation, 112(12), 1796–1808. https://doi.org/10.1172/jci200319246
Xu, H., Barnes, G. T., Yang, Q., Tan, G., Yang, D., Chou, C. J., et al. (2003). Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. The Journal of Clinical Investigation, 112(12), 1821–1830. https://doi.org/10.1172/jci200319451
Martinez, F. O., Sica, A., Mantovani, A., & Locati, M. (2008). Macrophage activation and polarization. Frontiers in Bioscience, 13, 453–461. https://doi.org/10.2741/2692
Kraakman, M. J., Murphy, A. J., Jandeleit-Dahm, K., & Kammoun, H. L. (2014). Macrophage polarization in obesity and type 2 diabetes: Weighing down our understanding of macrophage function? Frontiers in Immunology, 5, 470. https://doi.org/10.3389/fimmu.2014.00470
Kratz, M., Coats, B. R., Hisert, K. B., Hagman, D., Mutskov, V., Peris, E., et al. (2014). Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metabolism, 20(4), 614–625. https://doi.org/10.1016/j.cmet.2014.08.010
Xu, X., Grijalva, A., Skowronski, A., van Eijk, M., Serlie, M. J., & Ferrante, A. W., Jr. (2013). Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metabolism, 18(6), 816–830. https://doi.org/10.1016/j.cmet.2013.11.001
Hill, D. A., Lim, H. W., Kim, Y. H., Ho, W. Y., Foong, Y. H., Nelson, V. L., et al. (2018). Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proceedings of the National Academy of Sciences of the United States of America, 115(22), E5096–E5105. https://doi.org/10.1073/pnas.1802611115
Weinstock, A., Brown, E. J., Garabedian, M. L., Pena, S., Sharma, M., Lafaille, J., et al. (2019). Single-cell RNA sequencing of visceral adipose tissue leukocytes reveals that caloric restriction following obesity promotes the accumulation of a distinct macrophage population with features of phagocytic cells. Immunometabolism., 1, e190008. https://doi.org/10.20900/immunometab20190008
Lu, H., Clauser, K. R., Tam, W. L., Frose, J., Ye, X., Eaton, E. N., et al. (2014). A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nature Cell Biology, 16(11), 1105–1117. https://doi.org/10.1038/ncb3041
Jinushi, M., Chiba, S., Yoshiyama, H., Masutomi, K., Kinoshita, I., aka-Akita, H., et al. (2011). Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proceedings of the National Academy of Sciences of the United States of America, 108(30), 12425–30. https://doi.org/10.1073/pnas.1106645108
Zahorska-Markiewicz, B., Janowska, J., Olszanecka-Glinianowicz, M., & Zurakowski, A. (2000). Serum concentrations of TNF-alpha and soluble TNF-alpha receptors in obesity. International Journal of Obesity and Related Metabolic Disorders, 24(11), 1392–1395. https://doi.org/10.1038/sj.ijo.0801398
Fain, J. N., Bahouth, S. W., & Madan, A. K. (2004). TNFalpha release by the nonfat cells of human adipose tissue. International Journal of Obesity and Related Metabolic Disorders, 28(4), 616–622. https://doi.org/10.1038/sj.ijo.0802594
Weichhaus, M., Broom, I., & Bermano, G. (2011). The molecular contribution of TNF-α in the link between obesity and breast cancer. Oncology Reports, 25(2), 477–483. https://doi.org/10.3892/or.2010.1099
Hillers-Ziemer, L. E., McMahon, R. Q., Hietpas, M., Paderta, G., LeBeau, J., McCready, J., & Arendt, L. M. (2020). Obesity promotes cooperation of cancer stem-like cells and macrophages to enhance mammary tumor angiogenesis. Cancers (Basel)., 12, 502. https://doi.org/10.3390/cancers12020502
Bowers, L. W., Rossi, E. L., McDonell, S. B., Doerstling, S. S., Khatib, S. A., Lineberger, C. G., et al. (2018). Leptin signaling mediates obesity-associated CSC enrichment and EMT in preclinical TNBC models. Molecular Cancer Research, 16(5), 869–879. https://doi.org/10.1158/1541-7786.mcr-17-0508
Linde, N., Casanova-Acebes, M., Sosa, M. S., Mortha, A., Rahman, A., Farias, E., et al. (2018). Macrophages orchestrate breast cancer early dissemination and metastasis. Nature Communications, 9(1), 21. https://doi.org/10.1038/s41467-017-02481-5
Carron, E. C., Homra, S., Rosenberg, J., Coffelt, S. B., Kittrell, F., Zhang, Y., et al. (2017). Macrophages promote the progression of premalignant mammary lesions to invasive cancer. Oncotarget, 8(31), 50731–50746. https://doi.org/10.18632/oncotarget.14913
Chamberlin, T., Clack, M., Silvers, C., Kuziel, G., Thompson, V., Johnson, H., et al. (2020). Targeting obesity-induced macrophages during preneoplastic growth promotes mammary epithelial stem/progenitor activity, DNA damage, and tumor formation. Cancer Research, 80(20), 4465–4475. https://doi.org/10.1158/0008-5472.Can-20-0789
Muliaditan, T., Caron, J., Okesola, M., Opzoomer, J. W., Kosti, P., Georgouli, M., et al. (2018). Macrophages are exploited from an innate wound healing response to facilitate cancer metastasis. Nature Communications, 9(1), 2951. https://doi.org/10.1038/s41467-018-05346-7
Deligne, C., Murdamoothoo, D., Gammage, A. N., Gschwandtner, M., Erne, W., Loustau, T., et al. (2020). Matrix-targeting immunotherapy controls tumor growth and spread by switching macrophage phenotype. Cancer Immunology Research, 8(3), 368–382. https://doi.org/10.1158/2326-6066.Cir-19-0276
Catalán, V., Gómez-Ambrosi, J., Rodríguez, A., Ramírez, B., Rotellar, F., Valentí, V., et al. (2012). Increased tenascin C and Toll-like receptor 4 levels in visceral adipose tissue as a link between inflammation and extracellular matrix remodeling in obesity. Journal of Clinical Endocrinology and Metabolism, 97(10), E1880–E1889. https://doi.org/10.1210/jc.2012-1670
Lumeng, C. N., Bodzin, J. L., & Saltiel, A. R. (2007). Obesity induces a phenotypic switch in adipose tissue macrophage polarization. The Journal of Clinical Investigation, 117(1), 175–184. https://doi.org/10.1172/JCI29881
Zeyda, M., Farmer, D., Todoric, J., Aszmann, O., Speiser, M., Gyori, G., et al. (2007). Human adipose tissue macrophages are of an anti-inflammatory phenotype but capable of excessive pro-inflammatory mediator production. International Journal of Obesity, 31(9), 1420–1428. https://doi.org/10.1038/sj.ijo.0803632
Sun, X., Glynn, D. J., Hodson, L. J., Huo, C., Britt, K., Thompson, E. W., et al. (2017). CCL2-driven inflammation increases mammary gland stromal density and cancer susceptibility in a transgenic mouse model. Breast Cancer Research, 19(1), 4. https://doi.org/10.1186/s13058-016-0796-z
Kuziel, G., Thompson, V., D’Amato, J. V., & Arendt, L. M. (2020). Stromal CCL2 signaling promotes mammary tumor fibrosis through recruitment of myeloid-lineage cells. Cancers (Basel)., 12(8), 2083. https://doi.org/10.3390/cancers12082083
Burke, R. M., Madden, K. S., Perry, S. W., Zettel, M. L., & Brown, E. B., 3rd. (2013). Tumor-associated macrophages and stromal TNF-α regulate collagen structure in a breast tumor model as visualized by second harmonic generation. Journal of Biomedial Optics, 18(8), 86003. https://doi.org/10.1117/1.Jbo.18.8.086003
Piccinini, A. M., Zuliani-Alvarez, L., Lim, J. M., & Midwood, K. S. (2016). Distinct microenvironmental cues stimulate divergent TLR4-mediated signaling pathways in macrophages. Science Signaling, 9(443), ra86. https://doi.org/10.1126/scisignal.aaf3596
D’Esposito, V., Liguoro, D., Ambrosio, M. R., Collina, F., Cantile, M., Spinelli, R., et al. (2016). Adipose microenvironment promotes triple negative breast cancer cell invasiveness and dissemination by producing CCL5. Oncotarget, 7(17), 24495–24509. https://doi.org/10.18632/oncotarget.8336
Walens, A., DiMarco, A. V., Lupo, R., Kroger, B. R., Damrauer, J. S., & Alvarez, J. V. (2019). CCL5 promotes breast cancer recurrence through macrophage recruitment in residual tumors. eLife, 8, e43653. https://doi.org/10.7554/eLife.43653
Coffelt, S. B., Wellenstein, M. D., & de Visser, K. E. (2016). Neutrophils in cancer: Neutral no more. Nature Reviews Cancer, 16(7), 431–446. https://doi.org/10.1038/nrc.2016.52
Orlandini, L. F., Pimentel, F. F., Andrade, J. M., Reis, F., Mattos-Arruda, L., & Tiezzi, D. G. (2021). Obesity and high neutrophil-to-lymphocyte ratio are prognostic factors in non-metastatic breast cancer patients. Brazilian Journal of Medical and Biological Research, 54(10), e11409. https://doi.org/10.1590/1414-431X2021e11409
Strell, C., Lang, K., Niggemann, B., Zaenker, K. S., & Entschladen, F. (2010). Neutrophil granulocytes promote the migratory activity of MDA-MB-468 human breast carcinoma cells via ICAM-1. Experimental Cell Research, 316(1), 138–148. https://doi.org/10.1016/j.yexcr.2009.09.003
McDowell, S. A. C., Luo, R. B. E., Arabzadeh, A., Doré, S., Bennett, N. C., Breton, V., et al. (2021). Neutrophil oxidative stress mediates obesity-associated vascular dysfunction and metastatic transmigration. Nat Cancer., 2(5), 545–562. https://doi.org/10.1038/s43018-021-00194-9
Marvel, D., & Gabrilovich, D. I. (2015). Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. The Journal of Clinical Investigation, 125(9), 3356–3364. https://doi.org/10.1172/jci80005
Bronte, V., Brandau, S., Chen, S. H., Colombo, M. P., Frey, A. B., Greten, T. F., et al. (2016). Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nature Communications, 7, 12150. https://doi.org/10.1038/ncomms12150
Gabrilovich, D. I. (2017). Myeloid-derived suppressor cells. Cancer Immunology Research, 5(1), 3–8. https://doi.org/10.1158/2326-6066.Cir-16-0297
Okwan-Duodu, D., Umpierrez, G. E., Brawley, O. W., & Diaz, R. (2013). Obesity-driven inflammation and cancer risk: Role of myeloid derived suppressor cells and alternately activated macrophages. American Journal of Cancer Research, 3(1), 21–33.
Ostrand-Rosenberg, S. (2018). Myeloid derived-suppressor cells: Their role in cancer and obesity. Current Opinion in Immunology, 51, 68–75. https://doi.org/10.1016/j.coi.2018.03.007
Clements, V. K., Long, T., Long, R., Figley, C., Smith, D. M. C., & Ostrand-Rosenberg, S. (2018). Frontline Science: High fat diet and leptin promote tumor progression by inducing myeloid-derived suppressor cells. Journal of Leukocyte Biology, 103(3), 395–407. https://doi.org/10.1002/jlb.4hi0517-210r
Xia, S., Sha, H., Yang, L., Ji, Y., Ostrand-Rosenberg, S., & Qi, L. (2011). Gr-1+ CD11b+ myeloid-derived suppressor cells suppress inflammation and promote insulin sensitivity in obesity. Journal of Biological Chemistry, 286(26), 23591–23599. https://doi.org/10.1074/jbc.M111.237123
Gibson, J. T., Orlandella, R. M., Turbitt, W. J., Behring, M., Manne, U., Sorge, R. E., et al. (2020). Obesity-associated myeloid-derived suppressor cells promote apoptosis of tumor-infiltrating CD8 T cells and immunotherapy resistance in breast cancer. Frontiers in Immunology, 11, 590794. https://doi.org/10.3389/fimmu.2020.590794
Hossain, F., Al-Khami, A. A., Wyczechowska, D., Hernandez, C., Zheng, L., Reiss, K., et al. (2015). Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunology Research, 3(11), 1236–1247. https://doi.org/10.1158/2326-6066.Cir-15-0036
Al-Khami, A. A., Zheng, L., Del Valle, L., Hossain, F., Wyczechowska, D., Zabaleta, J., et al. (2017). Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunol., 6(10), e1344804. https://doi.org/10.1080/2162402x.2017.1344804
Macia, L., Delacre, M., Abboud, G., Ouk, T. S., Delanoye, A., Verwaerde, C., et al. (2006). Impairment of dendritic cell functionality and steady-state number in obese mice. The Journal of Immunology, 177(9), 5997–6006. https://doi.org/10.4049/jimmunol.177.9.5997
Ali, H. R., Provenzano, E., Dawson, S. J., Blows, F. M., Liu, B., Shah, M., et al. (2014). Association between CD8+ T-cell infiltration and breast cancer survival in 12,439 patients. Annals of Oncology, 25(8), 1536–1543. https://doi.org/10.1093/annonc/mdu191
Huang, Y., Ma, C., Zhang, Q., Ye, J., Wang, F., Zhang, Y., et al. (2015). CD4+ and CD8+ T cells have opposing roles in breast cancer progression and outcome. Oncotarget, 6(19), 17462–17478. https://doi.org/10.18632/oncotarget.3958
Floris, G., Richard, F., Hamy, A. S., Jongen, L., Wildiers, H., Ardui, J., et al. (2021). Body mass index and tumor-infiltrating lymphocytes in triple-negative breast cancer. Journal of the National Cancer Institute, 113(2), 146–153. https://doi.org/10.1093/jnci/djaa090
Takada, K., Kashiwagi, S., Asano, Y., Goto, W., Ishihara, S., Morisaki, T., et al. (2021). Clinical verification of body mass index and tumor immune response in patients with breast cancer receiving preoperative chemotherapy. Bmc Cancer, 21(1), 1129. https://doi.org/10.1186/s12885-021-08857-7
Wogsland, C. E., Lien, H. E., Pedersen, L., Hanjra, P., Grondal, S. M., Brekken, R. A., et al. (2021). High-dimensional immunotyping of tumors grown in obese and non-obese mice. Disease Models & Mechanisms, 14(4), dmm048977. https://doi.org/10.1242/dmm.048977
Neumann, S., Campbell, K., Woodall, M. J., Evans, M., Clarkson, A. N., & Young, S. L. (2021). Obesity has a systemic effect on immune cells in naïve and cancer-bearing mice. International Journal of Molecular Sciences, 22(16), 8803. https://doi.org/10.3390/ijms22168803
Baek, A. E., Yu, Y. A., He, S., Wardell, S. E., Chang, C. Y., Kwon, S., et al. (2017). The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nurse Aide Training Cetificate Community, 8(1), 864. https://doi.org/10.1038/s41467-017-00910-z
Nielsen, S. F., Nordestgaard, B. G., & Bojesen, S. E. (2012). Statin use and reduced cancer-related mortality. New England Journal of Medicine, 367(19), 1792–1802. https://doi.org/10.1056/NEJMoa1201735
Yang, H., Youm, Y. H., Vandanmagsar, B., Rood, J., Kumar, K. G., Butler, A. A., et al. (2009). Obesity accelerates thymic aging. Blood, 114(18), 3803–3812. https://doi.org/10.1182/blood-2009-03-213595
Yang, H., Youm, Y. H., Vandanmagsar, B., Ravussin, A., Gimble, J. M., Greenway, F., et al. (2010). Obesity increases the production of proinflammatory mediators from adipose tissue T cells and compromises TCR repertoire diversity: Implications for systemic inflammation and insulin resistance. The Journal of Immunology, 185(3), 1836–1845. https://doi.org/10.4049/jimmunol.1000021
Wherry, E. J., & Kurachi, M. (2015). Molecular and cellular insights into T cell exhaustion. Nature Reviews Immunology, 15(8), 486–499. https://doi.org/10.1038/nri3862
ElTanbouly, M. A., & Noelle, R. J. (2021). Rethinking peripheral T cell tolerance: Checkpoints across a T cell’s journey. Nature Reviews Immunology, 21(4), 257–267. https://doi.org/10.1038/s41577-020-00454-2
Strissel, K. J., DeFuria, J., Shaul, M. E., Bennett, G., Greenberg, A. S., & Obin, M. S. (2010). T-cell recruitment and Th1 polarization in adipose tissue during diet-induced obesity in C57BL/6 mice. Obesity (Silver Spring, Md)., 18(10), 1918–1925. https://doi.org/10.1038/oby.2010.1
Nishimura, S., Manabe, I., Nagasaki, M., Eto, K., Yamashita, H., Ohsugi, M., et al. (2009). CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nature Medicine, 15(8), 914–920. https://doi.org/10.1038/nm.1964
Porsche, C. E., Delproposto, J. B., Geletka, L., O’Rourke, R., & Lumeng, C. N. (2021). Obesity results in adipose tissue T cell exhaustion. JCI insight., 6(8), e139793. https://doi.org/10.1172/jci.insight.139793
Zhang, C., Yue, C., Herrmann, A., Song, J., Egelston, C., Wang, T., et al. (2020). STAT3 Activation-induced fatty acid oxidation in CD8(+) T effector cells is critical for obesity-promoted breast tumor growth. Cell Metabolism, 31(1), 148–61.e5. https://doi.org/10.1016/j.cmet.2019.10.013
Wang, J. C., Xu, Y., Huang, Z. M., & Lu, X. J. (2018). T cell exhaustion in cancer: Mechanisms and clinical implications. Journal of Cellular Biochemistry, 119(6), 4279–4286. https://doi.org/10.1002/jcb.26645
Kado, T., Nawaz, A., Takikawa, A., Usui, I., & Tobe, K. (2019). Linkage of CD8(+) T cell exhaustion with high-fat diet-induced tumourigenesis. Science and Reports, 9(1), 12284. https://doi.org/10.1038/s41598-019-48678-0
Castello, L. M., Raineri, D., Salmi, L., Clemente, N., Vaschetto, R., Quaglia, M., et al. (2017). Osteopontin at the crossroads of inflammation and tumor progression. Mediators of Inflammation, 2017, 4049098. https://doi.org/10.1155/2017/4049098
Beyer, M., Abdullah, Z., Chemnitz, J. M., Maisel, D., Sander, J., Lehmann, C., et al. (2016). Tumor-necrosis factor impairs CD4(+) T cell-mediated immunological control in chronic viral infection. Nature Immunology, 17(5), 593–603. https://doi.org/10.1038/ni.3399
Nave, H., Mueller, G., Siegmund, B., Jacobs, R., Stroh, T., Schueler, U., et al. (2008). Resistance of Janus kinase-2 dependent leptin signaling in natural killer (NK) cells: A novel mechanism of NK cell dysfunction in diet-induced obesity. Endocrinol., 149(7), 3370–3378. https://doi.org/10.1210/en.2007-1516
Viel, S., Besson, L., Charrier, E., Marçais, A., Disse, E., Bienvenu, J., et al. (2017). Alteration of natural killer cell phenotype and function in obese individuals. Clinical Immunology, 177, 12–17. https://doi.org/10.1016/j.clim.2016.01.007
Naujoks, W., Quandt, D., Hauffe, A., Kielstein, H., Bähr, I., & Spielmann, J. (2020). Characterization of surface receptor expression and cytotoxicity of human NK cells and NK cell subsets in overweight and obese humans. Frontiers in Immunology, 11, 573200. https://doi.org/10.3389/fimmu.2020.573200
Imai, K., Matsuyama, S., Miyake, S., Suga, K., & Nakachi, K. (2000). Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: An 11-year follow-up study of a general population. Lancet (London, England)., 356(9244), 1795–1799. https://doi.org/10.1016/s0140-6736(00)03231-1
Michelet, X., Dyck, L., Hogan, A., Loftus, R. M., Duquette, D., Wei, K., et al. (2018). Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nature Immunology, 19(12), 1330–1340. https://doi.org/10.1038/s41590-018-0251-7
Silva-Santos, B., Mensurado, S., & Coffelt, S. B. (2019). γδ T cells: Pleiotropic immune effectors with therapeutic potential in cancer. Nature Reviews Cancer, 19(7), 392–404. https://doi.org/10.1038/s41568-019-0153-5
Lopes, N., McIntyre, C., Martin, S., Raverdeau, M., Sumaria, N., Kohlgruber, A. C., et al. (2021). Distinct metabolic programs established in the thymus control effector functions of γδ T cell subsets in tumor microenvironments. Nature Immunology, 22(2), 179–192. https://doi.org/10.1038/s41590-020-00848-3
Lemoine, A. Y., Ledoux, S., & Larger, E. (2013). Adipose tissue angiogenesis in obesity. Thrombosis and Haemostasis, 110(4), 661–668. https://doi.org/10.1160/th13-01-0073
Mydlo, J. H., Gerstein, M. I., Harris, C. F., & Braverman, A. S. (2003). Immune function, mitogenicity, and angiogenic growth factor concentrations in lean and obese rodent sera: Implications in obesity-related prostate tumor biology. Prostate Cancer and Prostatic Diseases, 6(4), 286–289. https://doi.org/10.1038/sj.pcan.4500693
Ecker, B. L., Lee, J. Y., Sterner, C. J., Solomon, A. C., Pant, D. K., Shen, F., et al. (2019). Impact of obesity on breast cancer recurrence and minimal residual disease. Breast Cancer Research, 21(1), 41. https://doi.org/10.1186/s13058-018-1087-7
Wu, X., Zhang, X., Hao, Y., & Li, J. (2021). Obesity-related protein biomarkers for predicting breast cancer risk: An overview of systematic reviews. Breast Cancer, 28(1), 25–39. https://doi.org/10.1007/s12282-020-01182-0
Weidner, N., Folkman, J., Pozza, F., Bevilacqua, P., Allred, E. N., Moore, D. H., et al. (1992). Tumor angiogenesis: A new significant and independent prognostic indicator in early-stage breast carcinoma. Journal of the National Cancer Institute, 84(24), 1875–1887. https://doi.org/10.1093/jnci/84.24.1875
Weidner, N., Semple, J. P., Welch, W. R., & Folkman, J. (1991). Tumor angiogenesis and metastasis–Correlation in invasive breast carcinoma. New England Journal of Medicine, 324(1), 1–8. https://doi.org/10.1056/nejm199101033240101
Lim, H. Y., Seung, B. J., Cho, S. H., Kim, S. H., Bae, M. K., & Sur, J. H. (2022). Canine mammary cancer in overweight or obese female dogs is associated with intratumoral microvessel density and macrophage counts. Veterinary Pathology, 59(1), 39–45. https://doi.org/10.1177/03009858211040481
Gu, J.-W., Young, E., Patterson, S. G., Makey, K. L., Wells, J., Huang, M., et al. (2014). Postmenopausal obesity promotes tumor angiogenesis and breast cancer progression in mice. Cancer Biology & Therapy, 11(10), 910–917. https://doi.org/10.4161/cbt.11.10.15473
Cowen, S., McLaughlin, S. L., Hobbs, G., Coad, J., Martin, K. H., Olfert, I. M., et al. (2015). High-fat, high-calorie diet enhances mammary carcinogenesis and local inflammation in MMTV-PyMT mouse model of breast cancer. Cancers (Basel)., 7(3), 1125–1142. https://doi.org/10.3390/cancers7030828
Pelton, K., Coticchia, C. M., Curatolo, A. S., Schaffner, C. P., Zurakowski, D., Solomon, K. R., et al. (2014). Hypercholesterolemia induces angiogenesis and accelerates growth of breast tumors in vivo. American Journal of Pathology, 184(7), 2099–2110. https://doi.org/10.1016/j.ajpath.2014.03.006
Gonzalez-Perez, R. R., Lanier, V., & Newman, G. (2013). Leptin’s pro-angiogenic signature in breast cancer. Cancers (Basel)., 5(3), 1140–1162. https://doi.org/10.3390/cancers5031140
Incio, J., Ligibel, J. A., McManus, D. T., Suboj, P., Jung, K., Kawaguchi, K., et al. (2018). Obesity promotes resistance to anti-VEGF therapy in breast cancer by up-regulating IL-6 and potentially FGF-2. Science Translational Medicine, 10(432), eaag0945. https://doi.org/10.1126/scitranslmed.aag0945
Lin, E. Y., Li, J. F., Gnatovskiy, L., Deng, Y., Zhu, L., Grzesik, D. A., et al. (2006). Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Research, 66(23), 11238–11246. https://doi.org/10.1158/0008-5472.CAN-06-1278
De Palma, M., Venneri, M. A., Galli, R., Sergi Sergi, L., Politi, L. S., Sampaolesi, M., et al. (2005). Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell, 8(3), 211–226. https://doi.org/10.1016/j.ccr.2005.08.002
Arendt, L. M., McCready, J., Keller, P. J., Baker, D. D., Naber, S. P., Seewaldt, V., et al. (2013). Obesity promotes breast cancer by CCL2-mediated macrophage recruitment and angiogenesis. Cancer Research, 73(19), 6080–6093. https://doi.org/10.1158/0008-5472.can-13-0926
Yadav, N. V. S., Barcikowski, A., Uehana, Y., Jacobs, A. T., & Connelly, L. (2020). Breast adipocyte co-culture increases the expression of pro-angiogenic factors in macrophages. Frontiers in Oncology, 10, 454. https://doi.org/10.3389/fonc.2020.00454
Kolb, R., Phan, L., Borcherding, N., Liu, Y., Yuan, F., Janowski, A. M., et al. (2016). Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nature Communications, 7, 13007. https://doi.org/10.1038/ncomms13007
Kolb, R., Kluz, P., Tan, Z. W., Borcherding, N., Bormann, N., Vishwakarma, A., et al. (2019). Obesity-associated inflammation promotes angiogenesis and breast cancer via angiopoietin-like 4. Oncogene, 38(13), 2351–2363. https://doi.org/10.1038/s41388-018-0592-6
Gainsford, T., Willson, T. A., Metcalf, D., Handman, E., McFarlane, C., Ng, A., et al. (1996). Leptin can induce proliferation, differentiation, and functional activation of hemopoietic cells. Proceedings of the National Academy of Sciences of the United States of America, 93(25), 14564–14568. https://doi.org/10.1073/pnas.93.25.14564
McTiernan A. 2018 Weight, physical activity and breast cancer survival. Proc Nutr Soc.:1–9. doi:https://doi.org/10.1017/s0029665118000010.
Rossi, E. L., Dunlap, S. M., Bowers, L. W., Khatib, S. A., Doerstling, S. S., Smith, L. A., et al. (2017). Energy balance modulation impacts epigenetic reprogramming, ERalpha and ERbeta expression, and mammary tumor development in MMTV-neu transgenic mice. Cancer Research, 77(9), 2500–2511. https://doi.org/10.1158/0008-5472.can-16-2795
Pérez, L. M., Suárez, J., Bernal, A., de Lucas, B., San Martin, N., & Gálvez, B. G. (2016). Obesity-driven alterations in adipose-derived stem cells are partially restored by weight loss. Obesity (Silver Spring, Md)., 24(3), 661–669. https://doi.org/10.1002/oby.21405
Nitti, M. D., Hespe, G. E., Kataru, R. P., García Nores, G. D., Savetsky, I. L., Torrisi, J. S., et al. (2016). Obesity-induced lymphatic dysfunction is reversible with weight loss. Journal of Physiology, 594(23), 7073–7087. https://doi.org/10.1113/jp273061
Rossi, E. L., de Angel, R. E., Bowers, L. W., Khatib, S. A., Smith, L. A., Van Buren, E., et al. (2016). Obesity-associated alterations in inflammation, epigenetics, and mammary tumor growth persist in formerly obese mice. Cancer Prevention Research (Philadelphia, Pa.), 9(5), 339–348. https://doi.org/10.1158/1940-6207.capr-15-0348
Yam, C., Esteva, F. J., Patel, M. M., Raghavendra, A. S., Ueno, N. T., Moulder, S. L., et al. (2019). Efficacy and safety of the combination of metformin, everolimus and exemestane in overweight and obese postmenopausal patients with metastatic, hormone receptor-positive, HER2-negative breast cancer: A phase II study. Investigational New Drugs, 37(2), 345–351. https://doi.org/10.1007/s10637-018-0700-z
Esteva, F. J., Moulder, S. L., Gonzalez-Angulo, A. M., Ensor, J., Murray, J. L., Green, M. C., et al. (2013). Phase I trial of exemestane in combination with metformin and rosiglitazone in nondiabetic obese postmenopausal women with hormone receptor-positive metastatic breast cancer. Cancer Chemotherapy and Pharmacology, 71(1), 63–72. https://doi.org/10.1007/s00280-012-1977-9
Alsheikh, H. A. M., Metge, B. J., Ha, C. M., Hinshaw, D. C., Mota, M. S. V., Kammerud, S. C., et al. (2021). Normalizing glucose levels reconfigures the mammary tumor immune and metabolic microenvironment and decreases metastatic seeding. Cancer Letters, 517, 24–34. https://doi.org/10.1016/j.canlet.2021.05.022
Giles, E. D., Jindal, S., Wellberg, E. A., Schedin, T., Anderson, S. M., Thor, A. D., et al. (2018). Metformin inhibits stromal aromatase expression and tumor progression in a rodent model of postmenopausal breast cancer. Breast Cancer Research, 20(1), 50. https://doi.org/10.1186/s13058-018-0974-2
Goodwin, P. J., Dowling, R. J. O., Ennis, M., Chen, B. E., Parulekar, W. R., Shepherd, L. E., et al. (2021). Effect of metformin versus placebo on metabolic factors in the MA.32 randomized breast cancer trial. Npj breast cancer, 7(1), 74.
de Miranda, F. S., Guimarães, J. P. T., Menikdiwela, K. R., Mabry, B., Dhakal, R., Rahman, R. L., et al. (2021). Breast cancer and the renin-angiotensin system (RAS): Therapeutic approaches and related metabolic diseases. Molecular and Cellular Endocrinology, 528, 111245. https://doi.org/10.1016/j.mce.2021.111245
Chauhan, V. P., Chen, I. X., Tong, R., Ng, M. R., Martin, J. D., Naxerova, K., et al. (2019). Reprogramming the microenvironment with tumor-selective angiotensin blockers enhances cancer immunotherapy. Proceedings of the National Academy of Sciences of the United States of America, 116(22), 10674–10680. https://doi.org/10.1073/pnas.1819889116
Daquinag, A. C., Dadbin, A., Snyder, B., Wang, X., Sahin, A. A., Ueno, N. T., et al. (2017). Non-glycanated decorin is a drug target on human adipose stromal cells. Mol Ther Oncolytics., 6, 1–9. https://doi.org/10.1016/j.omto.2017.05.003
Daquinag, A. C., Tseng, C., Salameh, A., Zhang, Y., Amaya-Manzanares, F., Dadbin, A., et al. (2015). Depletion of white adipocyte progenitors induces beige adipocyte differentiation and suppresses obesity development. Cell Death and Differentiation, 22(2), 351–363. https://doi.org/10.1038/cdd.2014.148
Su F, Ahn S, Saha A, DiGiovanni J, Kolonin MG. 2018 Adipose stromal cell targeting suppresses prostate cancer epithelial-mesenchymal transition and chemoresistance. Oncogene. doi:https://doi.org/10.1038/s41388-018-0558-8
Qian, B. Z., Li, J., Zhang, H., Kitamura, T., Zhang, J., Campion, L. R., et al. (2011). CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature, 475(7355), 222–225. https://doi.org/10.1038/nature10138
Bonapace, L., Coissieux, M. M., Wyckoff, J., Mertz, K. D., Varga, Z., Junt, T., et al. (2014). Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature, 515(7525), 130–133. https://doi.org/10.1038/nature13862
MacDonald, K. P., Palmer, J. S., Cronau, S., Seppanen, E., Olver, S., Raffelt, N. C., et al. (2010). An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood, 116(19), 3955–3963. https://doi.org/10.1182/blood-2010-02-266296
Kumar, V., Donthireddy, L., Marvel, D., Condamine, T., Wang, F., Lavilla-Alonso, S., et al. (2017). Cancer-associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC infiltration of tumors. Cancer Cell, 32(5), 654–68.e5. https://doi.org/10.1016/j.ccell.2017.10.005
Kaneda, M. M., Cappello, P., Nguyen, A. V., Ralainirina, N., Hardamon, C. R., Foubert, P., et al. (2016). Macrophage PI3Kγ drives pancreatic ductal adenocarcinoma progression. Cancer Discovery, 6(8), 870–885. https://doi.org/10.1158/2159-8290.Cd-15-1346
Sharif, O., Brunner, J. S., Vogel, A., & Schabbauer, G. (2019). Macrophage rewiring by nutrient associated PI3K dependent pathways. Frontiers in Immunology, 10, 2002. https://doi.org/10.3389/fimmu.2019.02002
Borcherding, N., Kolb, R., Gullicksrud, J., Vikas, P., Zhu, Y., & Zhang, W. (2018). Keeping tumors in check: A mechanistic review of clinical response and resistance to immune checkpoint blockade in cancer. Journal of Molecular Biology, 430(14), 2014–2029. https://doi.org/10.1016/j.jmb.2018.05.030
Adams, S., Loi, S., Toppmeyer, D., Cescon, D. W., De Laurentiis, M., Nanda, R., et al. (2019). Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: Cohort B of the phase II KEYNOTE-086 study. Annals of Oncology, 30(3), 405–411. https://doi.org/10.1093/annonc/mdy518
Wang, Z., Aguilar, E. G., Luna, J. I., Dunai, C., Khuat, L. T., Le, C. T., et al. (2019). Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nature Medicine, 25(1), 141–151. https://doi.org/10.1038/s41591-018-0221-5
Naik, A., Monjazeb, A. M., & Decock, J. (2019). The obesity paradox in cancer, tumor immunology, and immunotherapy: Potential therapeutic implications in triple negative breast cancer. Frontiers in Immunology, 10, 1940. https://doi.org/10.3389/fimmu.2019.01940
Pingili, A. K., Chaib, M., Sipe, L. M., Miller, E. J., Teng, B., Sharma, R., et al. (2021). Immune checkpoint blockade reprograms systemic immune landscape and tumor microenvironment in obesity-associated breast cancer. Cell Reports, 35(12), 109285. https://doi.org/10.1016/j.celrep.2021.109285
Eun, Y., Kim, I. Y., Sun, J. M., Lee, J., Cha, H. S., Koh, E. M., et al. (2019). Risk factors for immune-related adverse events associated with anti-PD-1 pembrolizumab. Science and Reports, 9(1), 14039. https://doi.org/10.1038/s41598-019-50574-6
Acknowledgements
The authors would like to thank Katherine Lumsden, Brenna Walton, and Yue Xiong for critically reading the manuscript. BioRender was used to generate all figures present in this work.
Funding
This work was supported by NIH grants R01 CA227542 (L.M.A.) and F31 CA247265 (G.K.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was also supported in part by a grant to University of Wisconsin-Madison from the Howard Hughes Medical Institute through the James H. Gilliam Fellowships for Advanced Study program.
Author information
Authors and Affiliations
Contributions
LMA had the idea for the article. LEH-Z, GK, AEW, and BNM performed literature searches. LEH-Z drafted the manuscript, and all authors critically revised the work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hillers-Ziemer, L.E., Kuziel, G., Williams, A.E. et al. Breast cancer microenvironment and obesity: challenges for therapy. Cancer Metastasis Rev 41, 627–647 (2022). https://doi.org/10.1007/s10555-022-10031-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-022-10031-9