
Abstract
Purpose
The relationship of allergic diseases, such as asthma, hay fever, and eczema, with cancer is under debate. Observational studies have reported conflicting findings, but such studies are susceptible to confounding and reverse causation. Understanding the potential role of allergy in carcinogenesis may shed new light on the biological mechanisms underpinning intrinsic immunity and cancer.
Methods
We conducted a Mendelian randomization study, using germline genetic variants as instrumental variables, to determine the causal relevance of allergic disease and on two most common malignancies: breast cancer and prostate cancer. We used the summary statistics from the largest ever genome-wide association studies conducted on allergic disease (ncase = 180,129), asthma (ncase = 14,085), breast (ncase = 122,977), and prostate cancer (ncase = 79,148) and calculated odds ratios (ORs) and 95% confidence intervals (CIs) of cancer for allergic disease.
Results
We did not observe any evidence to support a causal association between allergic disease and risk of breast cancer overall [OR 1.00 (95% CI 0.96–1.04), p = 0.95] or by subtype (estrogen receptor (ER)+ [0.99 (0.95–1.04), p = 0.71], ER− [1.05 (0.99–1.10), p = 0.11]). We also did not find any evidence for an association with prostate cancer [1.00 (0.94–1.05), p = 0.93] or advanced subtype [0.97 (0.90–1.05), p = 0.46]. Sensitivity analyses did not reveal directional pleiotropy.
Conclusion
Our study does not support a causal effect of allergic disease on the risk of breast or prostate cancer. Future studies may be conducted to focus on understanding the causal role of allergic disease in cancer prognosis or drug responses (e.g., immunotherapy).
Similar content being viewed by others

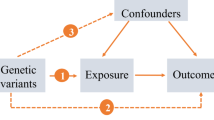

Introduction
There has been longstanding interest and debate on a possible relationship of allergic disease (e.g., asthma, hay fever, and eczema) with cancer [1, 2]. In general, two distinct and contradictory theories have been raised. The immune surveillance hypothesis proposes that the presence of allergies reflect an enhanced ability of the immune system to detect and destroy malignant cells; thus, having an allergic disorder may decrease a person’s cancer risk. In contrast, the antigen stimulation hypothesis suggests that chronic stimulation of the immune system by allergies may lead to increased levels of random pro-oncogenic mutations, repeated tissue inflammation, damage, and repair in favor of cancer onset [3]. Understanding the potential role of allergy in carcinogenesis is important as it may shed new light on the biological mechanisms underpinning intrinsic immunity and cancer and indicate therapeutic implications.
Epidemiologic evidence on associations of allergic disease with risk of breast and prostate cancer, the two most common hormone-driven malignancies among women and men, remains inconclusive [4,5,6,7,8]. Methodological concerns include small sample size, lack of proper control for confounding and bias. Meta-analyses provide a valuable way to evaluate the association combining results across studies. Vojtechova et al. aggregated data across 16 observational studies and found no evidence that allergy or asthma are associated with cancers of the breast (ncase = 10,736; relative risk (RR) [95% confidence interval (CI)] 1.01 [0.94–1.08] for any allergy; 0.93 [0.73–1.19] for asthma) or prostate (ncase = 7,890; 1.01 [0.87–1.17] for any allergy; 0.93 [0.76–1.15] for asthma) [9]. Similarly, Zhu et al. combined data from 20 observational studies and found no association of allergy or asthma with prostate cancer (ncase = 19,450; 0.96 [0.86–1.06] for any allergy; 1.04 [0.92–1.17] for asthma) [10]. Such findings, however, could have results due to attenuation to the null caused by measurement error or masking of a causal effect by confounders. For example, individuals with asthma or other allergic diseases might follow a different lifestyle than those without the diseases. They are likely to smoke less or refrain from certain foods to avoid irritation of diseases; they may pay more attention to environmental stimuli that could trigger their symptoms; they are less likely to engage with occupations that are exposed to high levels of chemical hazards. These confounders could possibly alter their risk of cancer onset.
Mendelian randomization (MR) overcomes the limitations of observational approaches by using genetic variants (mainly single nucleotide polymorphisms, SNPs) as instrumental variables (IVs) to evaluate the potential causal effect of an exposure on an outcome [11]. MR is based on the fact that: (i) SNPs (genotypes) are randomly assigned at conception, mirroring the randomization process in controlled trials and limiting the effect of confounding, and (ii) SNPs always precede disease onset, precluding reverse causality. Under certain assumptions, namely that the selected IVs are associated with the exposure, but not associated with any confounder of the exposure–outcome relationship, nor associated with the outcome via pathways other than through the exposure, an unconfounded estimate of causal effect of an exposure can be obtained using the observed IV-exposure and IV-outcome associations [12].
The rapidly increasing sample sizes of genome-wide association studies (GWAS) and the release of summary-level data, provides an unprecedented opportunity to conduct well-powered MR analyses. In this study, we conducted the first and also the largest two-sample MR analysis (with IV-exposure and IV-outcome associations from two different sets of participants) using genetic variants associated with a broad allergic disease phenotype including asthma, hay fever, or/and eczema as IVs, to appraise the causal relevance of allergic disease in cancers.
Methods
Data for IV-exposure
Two large-scale GWASs on allergic disease have been recently published. One was conducted by Ferreira et al. using 360,838 individuals of European ancestry which identified 136 independent risk variants associated with a self-reported broad allergic disease phenotype including asthma, hay fever (or allergic rhinitis), and eczema (or atopic dermatitis) with p < 310−8. [13] The other was conducted by Zhu et al. in 110,361 individuals of European ancestry and identified 38 independent risk variants associated with a self-reported doctor-diagnosed broad allergic disease phenotype including asthma, hay fever (or allergic rhinitis), and eczema with p < 510−8. [14] Zhu et al. also identified 32 asthma-associated SNPs and 33 other allergic diseases-associated SNPs.
For the current analysis, we retrieved summary data for the association between SNPs and allergic disease from both GWASs (136 + 38 = 174 SNPs). We filtered the 174 candidate variants on linkage disequilibrium (r2 < 0.20) and restricted to bi-allelic autosomal SNPs, from which, we obtained a total of 132 allergic disease-associated SNPs. Given the large overlap (> 90%) between participants of the two GWASs, and since a majority of final index SNPs (128 out of 132) were from Ferreira et al., we used effect sizes from the larger GWAS (Ferreira et al.) for index SNPs. We also included the 32 asthma-specific SNPs and 33 other allergic disease-associated SNPs from Zhu et al. in sensitivity analysis (the analytical procedure is presented in Fig. 1).

Flow chart of the current Mendelian randomization study. For the current analysis, we retrieved summary data for the association between SNPs and allergic disease from two GWASs of asthma and other allergic diseases (Ferreira et al. and Zhu et al.). We filtered these candidate variants on linkage disequilibrium (r2 < 0.20) and restricted to bi-allelic autosomal SNPs, from which, we obtained a total of 132 allergic disease-associated SNPs. Given the large overlap (> 90%) between the participants of the two GWASs, and since a majority of final index SNPs (128 out of 132) were from Ferreira et al., we used the effect sizes from the larger GWAS (Ferreira et al.) for those index SNPs. We performed a series of sensitivity analysis as well as used several MR approaches
Data for IV-outcome
We retrieved summary data for the association of allergic disease or asthma SNPs with breast and prostate cancer from the hitherto largest meta-GWAS of these outcomes conducted by the OncoArray network [15]. This is a large-scale collaborative effort established to understand the genetic architecture and biological mechanisms underlying five common cancers (breast, prostate, ovarian, colorectal, and lung). Participants of European ancestry were genotyped on a custom Illumina array and imputed to the 1000 Genomes Project reference panel. For each cancer type, results from individual participating GWAS were combined by fixed-effects inverse-variance weighted meta-analysis, with control for population stratification within each cohort and quality control thresholds of minor allele frequency > 0.01, imputation info score > 0.3, and Hardy–Weinberg equilibrium > 1 × 10–12 [16,17,18].
For breast cancer, 122,977 cases (105,974 controls) were involved, of which, 69,501 were estrogen receptor (ER)+ cases and 21,468 were ER− cases; for prostate cancer, 79,148 cases (61,106 controls) were involved, of which 15,167 were diagnosed with advanced prostate cancer (defined as Gleason score 8+ or prostate specific antigen [PSA] values > 100 ng/ml at diagnosis; or death from the disease or metastatic disease).
Statistical analysis
We conducted a two-sample MR to test for a potential causal relationship between allergic disease and risk of breast and prostate cancer and their subtypes. We applied a number of MR methods including a random-effects inverse-variance weighted approach (IVW) [19], a maximum likelihood method [20], MR-Egger regression [21], MR-PRESSO [22], and a weighted median approach [23].
For each of the k SNPs (IVs), the estimate of genetic association with exposure is represented as \(\hat{\beta }_{{{\text{Xk}}}}\) with standard error \(\sigma_{{{\text{Xk}}}}\) and the estimate of genetic association with outcome is represented as \(\hat{\beta }_{{{\text{Yk}}}}\) with standard error \(\sigma_{{{\text{Yk}}}}\). The IVW estimator can be interpreted as a weighted average of the ratio \(\frac{{\hat{\beta }_{{{\text{Yk}}}} }}{{\hat{\beta }_{{{\text{Xk}}}} }}\) for each variant, using the reciprocal of an approximate expression for their asymptotic variance \(\frac{{\sigma _{{{\text{Yk}}}}^{2} }}{{\hat{\beta }_{{{\text{Xk}}}}^{2} }}\). To evaluate potential heterogeneity among causal effects of different variants, we employed the Q-test. Complementary to IVW, we adopted the maximum likelihood method, which gives appropriately sized CIs when there is considerable imprecision in the estimates.
We performed MR-Egger regression and MR-PRESSO to both detect and correct for bias due to horizontal pleiotropy, where the average of direct effects of tested genetic variants on outcome is non-zero (i.e., violation of exclusion restriction assumption). Under the INstrument Strength Independent of Direct Effect (InSIDE) assumption, the intercept of MR-Egger regression of \(\hat{\beta }_{{{\text{Yk}}}}\) on \(\hat{\beta }_{{{\text{Xk}}}}\) will be different from zero in the presence of directional pleiotropy, and the slope of that regression will be a consistent estimate of the causal effect of X on Y.[24] In addition, MR-PRESSO implements a global test to evaluate the presence of specific outlier variants [22].
Finally, we employed a weighted median method that provides consistent estimates even when up to 50% of the analyzed genetic variants are invalid IVs.
Sensitivity analyses
We performed a series of sensitivity analyses. In our first sensitivity analysis, we restricted the IVs to SNPs at previous known loci and non-MHC regions. Thus, we used only validated results given that both GWASs lacked validations and excluded MHC region due to its complicated LD and pleiotropy with various diseases. In our second sensitivity analysis, we removed SNPs shown to be associated with potential confounders of the allergy–cancer association according to GWAS catalog (https://www.ebi.ac.uk/gwas/), such as depressive symptom measures, bone mineral density, alcohol consumption, markers of infection, and autoimmune diseases (Supplementary Table 1). In our third sensitivity analysis, we excluded one SNP at-a-time and performed IVW on the remaining 131 SNPs to identify potential influence of outlying variants on the estimates. Finally, we separated asthma from overall allergic disease phenotype to understand the cancer-specific effect of asthma.
We estimated the power of our study according to a method suggested by Brion et al. [25]. The 132 allergic disease-associated SNPs collectively explained 1.18% of the variance of the phenotype on the observed scale. We fixed the type-I error rate at 0.05.
Results
Under the current sample size (Table 1), our study had 80% power to detect a causal association of a relative 10.2% change in breast cancer risk and 12.8% for prostate cancer; corresponding estimates for ER+ breast cancer, ER− breast cancer, and advanced prostate cancer were 12.2%, 18.5%, and 12.1% relative changes. We also presented power estimations for a range of proportions of phenotype variation explained by the genetic variants (e.g., from 1 to 5%).
We did not find any evidence to support a causal role of allergic disease in overall (random-effects IVW OR [95% CI] 1.00 [0.96–1.04]), ER + (0.99 [0.95–1.04]), or ER− (1.05 [0.99–1.10]) breast cancer. Similarly, there was little evidence to support a causal role in overall prostate cancer (random-effects IVW OR [95% CI] 1.00 [0.94–1.05]) or its advanced subtype (0.97 [0.90–1.05]) (Table 2). Maximum likelihood methods provided similar results. However, we detected heterogeneity among the estimates of 132 index variants (all pheterogeneity < 0.001 for overall breast and prostate cancer, and their subtypes), indicating the existence of SNP-specific horizontal pleiotropy (scatter plots of effect sizes on exposure vs. outcome are shown in Supplementary Fig 1).
Estimates from MR-Egger and weighted median approach did not provide any evidence of an effect of allergic disease on breast or prostate cancer. We did not observe aggregated directional pleiotropy using MR-Egger (ppleiotropy for overall breast cancer, ER+ and ER− : 0.93, 1.00 and 0.85; ppleiotropy for overall prostate cancer and its advanced subtype: 0.96 and 0.89. Intercepts and 95% CIs are shown in Table 2). We further examined pleiotropy using MR-PRESSO. Although there appeared to be a few significant outliers in overall breast cancer and ER+ subset based on global test (none of the outliers were associated with known confounders), the outlier-corrected results were very similar to those from IVW for both breast (OR [95% CI] overall 1.02 [0.98–1.05]; ER+ 1.01 [0.97–1.05]; ER− 1.05 [0.99–1.10]) and prostate cancer (OR [95% CI] overall 1.00 [0.95–1.04]; advanced subtype 0.96 [0.90–1.04]).
The primary results did not substantially alter in our subsequent sensitivity analysis where we excluded SNPs from the HLA region (chr6: 25–35 Mb) and restricted to only known (and thus validated) SNPs (nSNP = 76; IVW OR [95% CI] for overall breast cancer 0.98 [0.93–1.04]; ER+ 0.98 [0.92–1.04]; ER− 1.04 [0.98–1.11]; for overall prostate cancer 0.99 [0.93–1.06]; advanced subtype 0.99 [0.90–1.09]) (Table 3). Consistent findings were observed when we excluded SNPs associated with potential confounders (nSNP = 107; IVW OR [95% CI] for overall breast cancer 0.98 [0.94–1.03]; ER+ 0.98 [0.93–1.03]; ER− 0.96 [0.90–1.02]; for overall prostate cancer 1.01 [0.95–1.08]; advanced subtype 0.99 [0.91–1.08]). Similar results were observed in the leave-one-out analysis where we iteratively removed one SNP each time and performed IVW using the remaining 131 SNPs (Supplementary Table 2). We did not find any causal effect of doctor-diagnosed asthma or other allergic disease on cancers across various MR approaches (Table 4).
Discussion
In this study, we used a strong instrumental variable based on 132 SNPs and capitalized on summary statistics of the largest meta-GWAS conducted for breast and prostate cancers in European populations. We aimed to determine whether the relationship between allergic disease and risk of two cancers was causal by using two-sample MR. In general, none of our analyses suggested a causal relationship between allergic disease, asthma, and breast or prostate cancer risk.
Our findings are in line with previous reports. Despite a few observational studies suggesting a possible association of allergic disease or asthma with breast or prostate cancer [26,27,28], such evidence has been scattered, restricted by small number of cancer cases (some studies included just a couple of hundred cases), and not supported by prospective epidemiological studies. For example, in the meta-analysis conducted by Vojtechova et al., no association was found for any allergy (RR [95% CI] 1.01 [0.94–1.08]) or asthma (0.93 [0.73–1.19]) with breast cancer, pooling results from both retrospective (ncase = 3,544) and prospective studies (ncase = 7,192). There was no difference in the effect estimates when cohort studies were analyzed separately [9]. Similarly, in the meta-analysis conducted by Zhu et al., restricting studies to those with a cohort design (ncase = 10,769), no association was observed for any allergy (1.06 [0.84–1.33]) or asthma (1.02 [0.91–1.15]) with prostate cancer [10]. Consistent with these previous findings, we did not observe evidence in support for an association between allergy, asthma, and risk of breast or prostate cancer.
The null findings of allergy with breast and prostate cancer are perhaps not surprising. Carcinogenesis is a multi-stage event where different defense mechanisms exist in different stages, including but not limited to, detoxification of metabolites originated from environmental carcinogens, decomposition to reactive oxygen species, DNA repair enzymes, and natural inhibitors of proliferating initiated cell [29]. The immune system, which acts as maybe the last line of host defense against cancer development, can be easily influenced by other existing defense mechanisms, leading to either surveillance or tolerance. In addition, the relation between allergy and cancer has been demonstrated complex and site-specific. While firm conclusions cannot be drawn for cancer at many sites, strong inverse associations have been reported for pancreatic cancer [30], glioma [31], and lymphoma [32], whereas positive association has been revealed for lung cancer [33] (although most such meta-analysis or pooled studies did not analyze prospective studies separately). Several shared biological mechanisms have been involved in the asthma–lung cancer association, including elevated levels of free radicals and reduced levels of antioxidants in the respiratory tract, persistent stimulation of cell regeneration to repair lung damage, and a heightened sensitivity to carcinogens [6]. While the global and individual mechanisms involved in other cancer sites might be more complicated and less straightforward than in the case of lung cancer, current evidence indicates an organ specific immunological effect elicited by allergic disease.
Our study has several strengths. We took a series of steps to guarantee the validity of estimates. We selected the most significant independent SNPs identified by the largest allergic disease GWAS, so all were robustly associated with the exposure of interest. These SNPs constitute a strong instrument with an F-statistic of 32.6. Secondly, none of the instrumental variables used in our analysis were cited by the NHGRI-EBI GWAS Catalog as associated with known strong confounders of cancer risk, such as BMI, smoking, or mammographic density at α = 10–8 level (a full list of confounders for asthma–breast cancer association includes weight, diet, exercise, alcohol consumption, smoking, stress and anxiety, levels of female hormone estrogen; while a full list of confounders is less clear for asthma–prostate cancer association, it probably includes diet, obesity, smoking, and chemical exposure). Nevertheless, some variants were indeed associated with autoimmune diseases, psychiatric traits and alcohol consumption; however, sensitivity analysis excluding those potentially confounding SNPs provided similar results as the primary analysis. Finally, we employed sensitivity analyses to control for pleiotropy and obtained highly consistent results.
Our current results also provide implications and directions for future research. While there is no strong evidence in support for a putative causal relationship between allergic disease, asthma, and the risk of cancers on breast and prostate, it is likely that the presence of allergies, cytokines, inflammatory, and immune responses it brings could influence the prognosis and mortality of cancer. For example, in a prospective cohort study of 1,102,247 cancer-free individuals at baseline, significant inverse associations between a history of both asthma and hay fever and overall cancer mortality (RR [95%CI] 0.88 [0.83–0.93]) as well as colorectal cancer mortality (0.76 [0.64–0.91]) were found. A history of hay fever only was associated with a significantly lowered risk of pancreatic cancer mortality, and a history of asthma only was associated with a significantly lowered risk of leukemia mortality [34]. Another study consisting of 475 incident pancreatic cancer cases found allergy to be associated with improved prognosis (longer survival among those with self-reported allergies than those without: 13.3 vs. 10.4 months) [35]. Future MR studies may be conducted to focus on understanding the causal role of allergic disease in cancer prognosis or drug responses (e.g., immunotherapy), rather than incidence. In addition, allergic disease belongs to type-I hypersensitivity, which is characterized by an atypical Th2-dominated immune response to innocuous environmental agents, leading to a large amount of IgE antibody production [36]. Most autoimmune inflammatory diseases such as systemic lupus erythematosus, rheumatoid arthritis and multiple sclerosis involve different immunological mechanisms such as the antigen–antibody complex triggered IgG involved type III hypersensitivity or cell-mediated type IV hypersensitivity. It is possible that these immune responses may be related to cancer development, and this therefore warrants further investigations.
Our study had also several caveats. Despite using the largest ever GWAS data on both the exposure and outcome, the statistical power in detecting a weak effect is poor—if the true causal effect of allergic disease on cancer was less than 5%, a magnitude that is probably of limited clinical importance—we only had power of 24% for breast cancer and 17% for prostate cancer, with our current sample size. Although we have involved clinically meaningful disease subtypes such as ER+ /− breast cancer and aggressive prostate cancer, we could not examine breast cancer based on menopause status (pre- and post-menopause; 85% of breast cancer cases in our samples are postmenopausal). In addition, for two-sample MR to be valid, the two samples have to be from the same underlying population, which is not the case for our sex-specific cancers. While our outcome data are breast cancer in female and prostate cancer in male, we could not examine the exposure trait stratified on sex; neither can we test the association of genetic instruments with other effect modifiers such as personal history of allergies, physical activity, and hormonal and lifestyle factors, due to lack of data. Violation of the exclusion restriction assumption may arise with a binary exposure in MR studies, if it is a dichotomization of a continuous risk factor (e.g., allergy could be a dichotomization of a continuous spectrum of subclinical problems), then the IVs can influence outcome via the continuous risk factor even if the binary exposure stays the same. In such cases, calculating a causal estimate can be difficult [37]. It may be helpful to test relevant continuous variables such as severity of allergy, and immunoglobulin E levels when GWAS of such phenotypes become available.
In conclusion, our MR analysis, despite its good overall statistical power and well-designed analytical protocol, provides no evidence in support of a causal relationship between allergic disease, asthma, and the risk of breast or prostate cancer.
References
Johnson KJ (1966) The relation of cancer to allergy. J Lancet 86:5–11
Shapiro S, Fedullo A (1973) Allergy and cancer. Lancet 301:1055–1056. https://doi.org/10.1016/S0140-6736(73)90686-7
Carrozzi L, Viegi G (2005) Allergy and cancer: a biological and epidemiological rebus. Allergy 60:1095–1097. https://doi.org/10.1111/j.1398-9995.2005.00940.x
Wang H, Diepgen TL (2005) Is atopy a protective or a risk factor for cancer? A review of epidemiological studies. Allergy 60:1098–1111. https://doi.org/10.1111/j.1398-9995.2005.00813.x
Rittmeyer D, Lorentz A (2012) Relationship between allergy and cancer: an overview. Int Arch Allergy Immunol 159:216–225. https://doi.org/10.1159/000338994
Turner MC, Chen Y, Krewski D, Ghadirian P (2006) An overview of the association between allergy and cancer. Int J Cancer 118:3124–3132. https://doi.org/10.1002/ijc.21752
Merrill RM, Isakson RT, Beck RE (2007) The association between allergies and cancer: what is currently known? Ann Allergy Asthma Immunol 99:102–116 (quiz 117–119, 150). https://doi.org/10.1016/S1081-1206(10)60632-1
Turner MC (2012) Epidemiology: allergy history, IgE, and cancer. Cancer Immunol Immunother CII 61:1493–1510. https://doi.org/10.1007/s00262-011-1180-6
Vojtechova P, Martin RM (2009) The association of atopic diseases with breast, prostate, and colorectal cancers: a meta-analysis. Cancer Causes Control CCC 20:1091–1105. https://doi.org/10.1007/s10552-009-9334-y
Zhu J, Song J, Liu Z et al (2016) Association between allergic conditions and risk of prostate cancer: a prisma-compliant systematic review and meta-analysis. Sci Rep 6:35682. https://doi.org/10.1038/srep35682
Davey Smith G, Ebrahim S (2003) ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 32:1–22. https://doi.org/10.1093/ije/dyg070
VanderWeele TJ, Tchetgen Tchetgen EJ, Cornelis M, Kraft P (2014) Methodological challenges in mendelian randomization. Epidemiol Camb Mass 25:427–435. https://doi.org/10.1097/EDE.0000000000000081
Ferreira MA, Vonk JM, Baurecht H et al (2017) Shared genetic origin of asthma, hay fever and eczema elucidates allergic disease biology. Nat Genet 49:1752–1757. https://doi.org/10.1038/ng.3985
Zhu Z, Lee PH, Chaffin MD et al (2018) A genome-wide cross-trait analysis from UK Biobank highlights the shared genetic architecture of asthma and allergic diseases. Nat Genet 50:857–864. https://doi.org/10.1038/s41588-018-0121-0
Amos CI, Dennis J, Wang Z et al (2016) The ONCOARRAY consortium: a network for understanding the genetic architecture of common cancers. Cancer Epidemiol Biomark Prev Publ. https://doi.org/10.1158/1055-9965.EPI-16-0106
Michailidou K, Lindström S, Dennis J et al (2017) Association analysis identifies 65 new breast cancer risk loci. Nature. https://doi.org/10.1038/nature24284
Milne RL, Kuchenbaecker KB, Michailidou K et al (2017) Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer. Nat Genet. https://doi.org/10.1038/ng.3785
Schumacher F, Al Olama A, Berndt S (2018) Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat Genet 50:928–936
Burgess S, Scott RA, Timpson NJ et al (2015) Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol 30:543–552. https://doi.org/10.1007/s10654-015-0011-z
Burgess S, Butterworth A, Thompson SG (2013) Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol 37:658–665. https://doi.org/10.1002/gepi.21758
Bowden J, Davey Smith G, Burgess S (2015) Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 44:512–525. https://doi.org/10.1093/ije/dyv080
Verbanck M, Chen C-Y, Neale B, Do R (2018) Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet 50:693–698. https://doi.org/10.1038/s41588-018-0099-7
Bowden J, Davey Smith G, Haycock PC, Burgess S (2016) Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 40:304–314. https://doi.org/10.1002/gepi.21965
Burgess S, Thompson SG (2017) Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol 32:377–389. https://doi.org/10.1007/s10654-017-0255-x
Brion M-JA, Shakhbazov K, Visscher PM (2013) Calculating statistical power in Mendelian randomization studies. Int J Epidemiol 42:1497–1501. https://doi.org/10.1093/ije/dyt179
Severi G, Baglietto L, Muller DC et al (2010) Asthma, asthma medications, and prostate cancer risk. Cancer Epidemiol Biomark Prev Publ 19:2318–2324. https://doi.org/10.1158/1055-9965.EPI-10-0381
Lowcock EC, Cotterchio M, Ahmad N (2013) Association between allergies, asthma, and breast cancer risk among women in Ontario, Canada. Cancer Causes Control CCC 24:1053–1056. https://doi.org/10.1007/s10552-013-0177-1
Hedderson MM, Malone KE, Daling JR, White E (2003) Allergy and risk of breast cancer among young women (United States). Cancer Causes Control CCC 14:619–626
Nakachi K, Hayashi T, Imai K, Kusunoki Y (2004) Perspectives on cancer immuno-epidemiology. Cancer Sci 95:921–929
Olson SH, Hsu M, Satagopan JM et al (2013) Allergies and risk of pancreatic cancer: a pooled analysis from the Pancreatic Cancer Case-Control Consortium. Am J Epidemiol 178:691–700. https://doi.org/10.1093/aje/kwt052
Linos E, Raine T, Alonso A, Michaud D (2007) Atopy and risk of brain tumors: a meta-analysis. J Natl Cancer Inst 99:1544–1550. https://doi.org/10.1093/jnci/djm170
Vajdic CM, Falster MO, de Sanjose S et al (2009) Atopic disease and risk of non-Hodgkin lymphoma: an InterLymph pooled analysis. Cancer Res 69:6482–6489. https://doi.org/10.1158/0008-5472.CAN-08-4372
Santillan AA, Camargo CA, Colditz GA (2003) A meta-analysis of asthma and risk of lung cancer (United States). Cancer Causes Control CCC 14:327–334
Turner MC, Chen Y, Krewski D et al (2005) Cancer mortality among US men and women with asthma and hay fever. Am J Epidemiol 162:212–221. https://doi.org/10.1093/aje/kwi193
Olson SH, Chou JF, Ludwig E et al (2010) Allergies, obesity, other risk factors and survival from pancreatic cancer. Int J Cancer 127:2412–2419. https://doi.org/10.1002/ijc.25240
Knopf PM (2000) Immunomodulation and allergy. Allergy Asthma Proc 21:215–220
Burgess S, Labrecque JA (2018) Mendelian randomization with a binary exposure variable: interpretation and presentation of causal estimates. Eur J Epidemiol 33:947–952. https://doi.org/10.1007/s10654-018-0424-6
Acknowledgments
Open access funding provided by Karolinska Institute. The breast cancer genome-wide association analyses were supported by the Government of Canada through Genome Canada and the Canadian Institutes of Health Research, the ‘Ministère de l’Économie, de la Science et de l’Innovation du Québec’ through Genome Québec and grant PSR-SIIRI-701, The National Institutes of Health (U19 CA148065, X01HG007492), Cancer Research UK (C1287/A10118, C1287/A16563, C1287/A10710), and The European Union (HEALTH-F2-2009-223175 and H2020 633784 and 634935). All studies and funders are listed in Michailidou et al. (2017).
Funding
This work was supported by the Veteskapsrådet International Postdoc grant (XJ); CB is supported by a Universidade de São Paulo/ Coordenação de Aperfeiçoamento de Pessoal de Nível Superior fellowship (processo no. 88887.160006/2017-00). The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health. Where authors are identified as personnel of the International Agency for Research on Cancer/World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy, or views of the International Agency for Research on Cancer/World Health Organization.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Members from the PRACTICAL, CRUK, BPC3, CAPS, and PEGASUS consortia are provided in the Supplement Notes. Information of the PRACTICAL consortium can be found at http://practical.icr.ac.uk/.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jiang, X., Dimou, N.L., Zhu, Z. et al. Allergy, asthma, and the risk of breast and prostate cancer: a Mendelian randomization study. Cancer Causes Control 31, 273–282 (2020). https://doi.org/10.1007/s10552-020-01271-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10552-020-01271-7