
Abstract
Objective
To assess the expression of rabies virus G-glycoprotein (RVGP) expression using Semliki Forest virus as a vector in combination with BHK-21 cells cultured in suspension.
Results
A multilevel factorial design was used to quantify effects of temperature (33–37 °C), fresh medium addition after the viral adsorption step (100–200 % with respect to the initial cell suspension volume before infection) and harvest time (8–40 h) on RVGP production. Experimental runs were performed in 24-well cell culture plates at a multiplicity of infection (MOI) of 16. An additional experiment in spinner-flask was performed at MOI of 9, using the optimal conditions determined in cell culture plates. Values for temperature, fresh medium addition and harvest time of 33 °C, 100 % and 16 h, respectively, ensured the optimal RVGP production in culture plates. The volumetric yield (239 ng ml−1) in these conditions was higher than that reported previously for adherent cell culture. In spinner-flasks, the volumetric yield was improved (559 ng ml−1).
Conclusion
These results establish the basis for designing bioprocess to produce RVGP.
Similar content being viewed by others
Introduction
Mammalian cells have become the host of choice for the production of recombinant glycoproteins for medical and veterinarian as well as structural biology applications (Baldi et al. 2007), primarily due to the need for post-translational modifications in order to guarantee the full bioactivity of these macromolecules (Walsh 2010). Most recombinant glycoproteins have been expressed in rodent cell lines such as Chinese hamster ovary (CHO), baby hamster kidney (BHK) and mouse myeloma (NS0) cells. These cell lines can be cultured in suspension. Specifically, the BHK-21 cell line has also been employed extensively for veterinarian viral vaccines (foot-and-mouth disease virus and rabies virus) since the 1960s (Auninš 2010).
More than half of biopharmaceutical products on the market are glycosylated proteins (Zhu 2012). Currently, there are two established strategies in the biopharmaceutical industry and academia to produce heterologous proteins: the use of stable cell lines, or transient gene expression. Transient gene expression offers the advantage of shorter development times, leading to much lower development costs when compared to stable cell line development. Furthermore, the quality of the products obtained from this strategy is suitable for preclinical assessment, thus expediting the “Proof-of-Principle” stage prior to advancing into the formal development process (Zhu 2012). In transient gene expression, non-viral (plasmid vectors) and viral expression vectors have been designed for transferring genes to mammalian cells (Baldi et al. 2007). In the past, the volumetric productivities reached by transient gene expression were not competitive with those from the stable cell lines. However, more recently, productivity up to 1 g/l for monoclonal antibodies has been reached in HEK-293E and CHO cell lines using non-viral vectors (Zhu 2012). These reports encourage further studies on transient gene expression in order to establish these processes at higher volumetric scales (100–1000 l range).
Approximately 55,000 people around the globe, mostly children, die each year of rabies, making human rabies a significant public health problem (Zhang et al. 2013). The approved licensed vaccines for human and animal use against rabies virus are inactivated whole virus vaccines with the exception of the canarypox-virus vectored vaccine against feline rabies (PUREVAX® Feline Rabies and PUREVAX® Rabies, Merial) (Jas et al. 2012). Subunit vaccines for rabies virus are in the development phase, and they are mostly based on G-glycoprotein. Its antigenicity has been extensively studied and exploited in vaccine development in the past few decades (Benmaamar et al. 2009). Animal cells as well as transgenic plants have been the most frequently used hosts for G-glycoprotein expression using both stable and transient gene expression (Benmaamar et al. 2009; Ventini et al. 2010; Loza-Rubio et al. 2012).
Semliki Forest virus (SFV) is a single positive-strand RNA virus with an envelope structure. Its genome has been introduced into two plasmid vectors as cDNA copies. The expression vector contains the SFV nonstructural genes (nsP1-4), the strong subgenomic SFV 26S promoter and a multilinker cloning region to introduce foreign genes for recombinant protein expression. The SFV structural proteins are provided from the helper vector, including the envelope and capsid proteins. On a laboratory scale, the SFV vector has been widely used for obtaining sufficient amounts of recombinant proteins, mainly functional membrane proteins, receptors, enzymes and viral proteins (Benmaamar et al. 2009), for cellular, or molecular assays, pharmacological tests and structural studies (Cabaniols et al. 2009). High volumetric protein expression levels and yields have been obtained using a modified SFV in mammalian cells, up to 10 mg L−1 and 100–300 pmol mg−1 of total proteins, for G protein-coupled receptors (Sevastsyanovich et al. 2009). Among the most widely used and assessed hosts in transient protein expression with SFV vector is BHK-21 cell line (Andréll and Tate 2013).
The aim of this work was to assess the rabies virus G-Glycoprotein (RVGP) expression using SFV as a vector in combination with the BHK-21 cell line cultured in suspension systems.
Materials and methods
Animal cell culture for SFV-RVGP infection
BHK-21 (C-13) cells (Sigma-Aldrich ECACC Cell Lines, Lyon, France) adapted to single cell suspension culture were kindly supplied by Dr. Renaud Wagner from Ecole Superieure Biotecnologie de Strasbourg (France) and stored in liquid nitrogen. The culture medium used had the following composition by volume: Iscove´s Modified Dulbecco Medium with glutamine and Phenol red (IMDM, catalog number 12200-036, Gibco, New York, USA) 45.5 %; High glucose Dulbecco´s Modified Eagle Medium (DMEM, catalog number 12100-046, Gibco, New York, USA) 45.5 %; heat inactivated fetal bovine serum (Catalog number 10082-147, Gibco, New York, USA) 5; 10 % m/v Pluronic F-68 (Sigma-Aldrich, St. Louis, USA) aqueous solution 2 %; and 4 mM glutamine (Sigma-Aldrich, St. Louis, USA) aqueous solution 2 %.
One milliliter of BHK-21 cells (2 × 106 cell ml−1) was thawed and placed in a 75 cm2 tissue culture flask (vertical position) with 30 ml of culture medium for growing. Four days later, the cell suspension was used to inoculate (2.5 × 05 cell ml−1) another 75 cm2 tissue culture flask, and enough culture medium was added to reach final cellular suspensions of 15 ml. Cells were cultured for 72 h. This cell suspension was used to inoculate (2.5 × 105 cell ml−1) 100 ml spinner flasks with 50 ml working volume at 60 rpm (Sci-Era quad drive stirrer system with a stirrer, Bellco Biotechnology, Vineland, USA). Cells in spinner flasks were cultured for 48 h (at which time the cell concentration was 2 × 106 cells ml−1), and then used for cell infection with recombinant SFV both in 24 well cell culture plates (Techno Plastic Products AG; TPP, Trasadingen, Switzerland) and spinner flask. The cell cultures in static and agitated systems were carried out at 37 °C and 5 % CO2 (Thermoforma 3110, Marietta, USA).
Cell concentration and viability. Specific cell death rate
The cells were counted using a Neubauer improved counting chamber (Precicolor HBG, Gießen, Germany) with proper sample dilution using Phosphate Buffered Saline. In parallel, viable cells were quantified using the trypan blue exclusion method (Augusto et al. 2010). The specific cell death rate, Kd, was calculated by plotting the natural logarithm of viable cell concentration versus time during the cell death phase.
Glutamine quantification
Glutamine concentrations from supernatant samples were measured using enzyme-coupled reaction and electrochemical detection, in YSI 2700 Select Bioanalyzer (YSI Life Sciences, Yellow Springs, USA).
PCR amplification and molecular cloning. In vitro-transcription and recombinant SFV-RVGP production
The PCR amplification and molecular cloning for expression (pSFV-RVGP) and helper (pSFV-Helper) plasmids as well as DNA propagation and linearization were described previously in detail (Benmaamar et al. 2009).
In vitro-transcription was performed after plasmid linearization, using MAXIScript® SP6 kit (Ambion, Austin, USA) according to manufacturer instructions with some modifications, such as: incubation temperature (40 °C) and transcription time (2 h) as well as the addition of two reagents, CAP analog (Invitrogen, Carlsbad, USA) and Ribolock (Fermentas, Vilnius, Lithuania). RNA quantification was performed by fluorometric method (Qubit, Invitrogen, Carlsbad, USA). Transcription efficiency was verified by electrophoresis in 0.8 % agarose gel.
RNA transfection by electroporation (GenePulser II, Biorad, Munich, Germany) to adherent BHK-21 C-13 cells (Adolfo Lutz Institute, São Paulo, Brazil) was carried out according to a defined procedure (Karlsson and Liljestrom 2003) with some modifications. RNAs recently obtained are transfected to cells. BHK-21 cells were cultivated in 75 cm2 T-flasks until approximately 80 % confluence. The ratio of pSFV-RVGP RNA to pSFV-Helper RNA was 2. The electroporation parameters were, 360 V, 75 µF; Resistance (∞), cuvettes with 0.2 cm gap width. Electroporated cells were cultured in 75 cm2 T-flasks for 24 h using 10 ml of DMEM medium supplemented with 10 % fetal bovine serum, at 37 °C in 5 % CO2 atmosphere. Subsequently, supernatant with recombinant virus from cell culture was collected by centrifugation and stored at -80 °C until cell infection or quantification.
Recombinant virus (SFV-RVGP) titration was defined by quantitative real time Polymerase Chain Reaction (qPCR) in StepOne Real Time PCR System (Applied Biosystem, Foster City, USA), RNA extraction, treatment with deoxyribonuclease and reverse transcription were performed as described previously (Puglia et al. 2013).
Cell infection
The produced SFV-RVGP (8 × 107 virus particles ml−1) was activated using 36 µL α-chymotrypsin (Sigma-Aldrich, St. Louis, MO, USA) aqueous solution (40 mg ml−1) for 30 min. Then, α-chymotrypsin was inactivated with addition of 100 µL aprotinin (Sigma-Aldrich, St. Louis, MO, USA) aqueous solution (10 mg ml−1) for 5 min. The volumes of α-chymotrypsin and aprotinin aqueous solutions are referred to one milliliter of inactivated SFV-RVGP. Both enzymatic reactions were performed at room temperature (24 °C) without agitation. Activated recombinant SFV-RVGP was prepared immediately before use.
Multifactorial experimental design was proposed to explore the experimental domain defined by temperature (33 and 37 °C), fresh culture medium addition after virus adsorption (100 and 200 % with respect to initial cell suspension volume) and harvest time (8, 16, 24, 32 and 40 h) on RVGP production in nanograms. All level combinations of three parameters under consideration were performed in triplicate; a single control experiment without recombinant virus addition was also executed. Therefore, each 24-well cell culture plate was used efficiently. The multiplicity of infection (ratio of recombinant virus to cell) for this experimental approach was 16. This study was carried out in 24 well cell culture plates. Two additional samples at 0 and 48 h post-infection, not included in experimental design, were also collected for each combination between temperature and fresh culture medium addition after virus adsorption levels in order to study expression profiles in more detail. For cell infection in different experimental conditions, 0.5 ml of cell suspension from spinner flask were plated per well. An activated recombinant virus amount to ensure the proper multiplicity of infection value was added immediately. The step of viral adsorption was performed at 150 rpm (Orbital Titer Plate Shaker, IKA MTS 2/4 DS1, Staufen, Germany), 37 °C and 5 % CO2 atmosphere for 120 min. Then, fresh culture medium volume was added and temperature was set according to specific conditions for each run included in multifactorial design. Samples were collected every 8 h. A portion of the sample was used to determine cell concentration and viability. For the remaining sample, cell and culture medium were separated by centrifugation at 750×g, 4 °C, for 4 min (Sorvall Biofuge Primo R Centrifuge, Thermo Electron, Langenselbold, Germany). Both types of samples were stored at −20 °C until RVGP and glutamine quantification.
For cell infection experiment performed in spinner flask, culture conditions before infection and recombinant virus activation procedure were similar to those described previously for experiments in cell culture plates. After 48 h in culture, 59 % of the BHK-21 broth volume in spinner flask was removed. Then, an activated virus volume to ensure MOI of 9 was added to the remaining broth volume. Virus adsorption was performed during 120 min at 37 °C. Subsequently, fresh culture medium volume was placed to restore the initial working volume in spinner flask (50 ml). Afterwards, temperature was diminished to 33 °C in 15 min, and after that, it was kept constant. RVGP production, cell concentration and viability were monitored for 48 h at 8 h intervals.
Enzyme-linked immunosorbent assay
The expression of RVGP, from culture medium and cells, was quantified by enzyme-linked immunosorbent assay (ELISA). The specific ELISA was performed according to instructions from the Rabies Virus Glycoprotein Enzyme Immunoassay Kit (Institut Pasteur, Paris, France). Details about sample preparations and procedure were reported previously (Benmaamar et al. 2009).
Statistical analysis
Experimental schedule and data analysis associated to multilevel factorial design was performed in Statgraphics Plus 5.0 (Statistical Graphics, Fairfax, USA). In order to determine the significant factors in adjusted polynomial model was used Pareto chart. The individual effect of each factor in range under study on RVGP production was shown graphically (main effect plot). The response surface for RVGP production was represented in tridimensional graph, keeping constant, one of the three considered factors. The statistical significance level (α) for decisions was 0.05. The other graphic data treatments were carried out in Microsoft Excel (Microsoft Office 2010, Microsoft, Redmond, USA).
Results
Experiments performed in cell culture plates
In every assessed experimental combination of temperature and fresh culture medium addition after virus adsorption step, a stop in growth and decrease in viability were detected in cells expressing RVGP when compared to the control experiment (cells non infected by SFV-GPV) (Fig. 1). These phenomena were observed earlier, firstly in cell infections performed at 33 °C. Nevertheless, taking into account the specific cell death rate [Kd], the cell death was sharper at 37 °C (Fig. 1; Table 1).

Viable cell concentration, cell viability (a–d) and RVGP expression and glutamine consumption profiles (e–h) for cell infections performed at 37 °C-100 % fresh culture medium addition, 37 °C-200 % fresh culture medium addition, 33 °C-100 % fresh culture medium addition, 33 °C-200 % fresh culture medium addition, respectively. Each point represents the average from three values, whereas points associated to control experiments represent a single sample. Error bars in RVGP expression and glutamine consumption profiles represent standard deviation, the considered time corresponds to viral post-infection time. The results are derived from 24-well cell culture plates
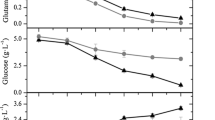
Diminution of temperature after cell infection was advantageous for RVGP expression (Fig. 1, 2). Among three under consideration, temperature (A) was the only parameter with individual statistical significance on RVGP production (Fig. 2a). The influence of post-infection fresh culture medium addition (B) and harvest time (C) were significant as interactions, B–C and A–B (Fig. 2a). The B–C interaction had a positive influence on RVGP production, this means that at higher levels of post-infection fresh culture medium addition and time, high values of RVGP production could be expected. However, the A-B interaction showed a negative influence on RVGP production, this suggests that combinations of higher with lower levels of these parameters could improve RVGP production. For instance, when temperature is low (33 °C), the fresh medium addition should be high (200 %) in order to obtain an improved production of RVGP. Post-infection or harvest time showed a nonlinear relationship with respect to RVGP concentration (Fig. 1e–h) and production (Fig. 2b), even without statistical significance. The maximum RVGP expression was reached at 16 h post-infection in four assessed combinations of temperature and fresh medium addition. After 16 h post-infection, as a rule, a decrease was observed in RVGP concentration. However, the experimental combination of 33 °C temperature and 200 % fresh medium addition showed no RVGP concentration decrease after 16 h post-infection (Fig. 1e–h). Taking into account 16 h as a reference to compare the impact of temperature on RVGP concentration, 54.2 % (Fig. 1e, g) and 53.0 % (Fig. 1f, h) increase were observed for 100 and 200 % fresh medium addition, respectively, when the temperature decreased by 4 °C (from 37 to 33 °C).

a Standardized Pareto chart for individual factors, two-factors combinations and quadratic term for harvest time, influencing RVGP production (ng) in experiments performed in 24-well cell culture plates according a multilevel factorial design. b Main effects plot for RVGP production (ng). c Estimated response surface for RVGP production (ng). Fresh culture medium addition after virus adsorption was kept constant (100 %). The variables: temperature, harvest time and fresh culture medium addition are represented as a, b and c, respectively
The selection of optimal harvest time was critical for experiments performed at 37 °C–200 % fresh culture medium addition and 33 °C–100 % fresh culture medium addition because of the significant decline of RVGP concentration, after maximum concentration point (16 h). For the remaining two combinations, the tolerance for harvesting was higher (Fig. 1e–h).
The increase of fresh culture medium addition from 100 to 200 % with respect to the initial cell suspension was not significant in order to improve RVGP production (Fig. 2a, b). The glutamine in culture medium was not totally consumed in any case. The consumption of this nutrient by cells at 37 °C was higher than 33 °C. However, at the end of cell infection period, the remaining glutamine in culture medium was always higher than 38.5 % with respect to its initial concentration (Fig. 1e–h).
Taking into consideration all findings described above, the best conditions for obtaining maximum RVGP production were 33 °C post-viral adsorption temperature, 100 % fresh culture medium addition and 16 h harvest time (Fig. 2c).
The RVGP expression is mainly distributed in the cell rather than supernatant fraction. There exists a tendency to increase the RVGP percentage in supernatant over the course of assessed infection time for experiments performed at 33 °C. This was not clearly observed for experiments at 37 °C (Fig. 3). For optimal RVGP expression condition, the percentage of RVGP in cells was 94.1 % (Fig. 3c). Experiments carried out at 33 °C showed RVGP expressions higher than 330 ng/106 cells (Fig. 3c, d).

RVGP expression kinetic, protein production is referred per 106 cells and RVGP distribution in supernatant and cells for cell infections at 37 °C-100 % fresh culture medium addition (a), 37 °C-200 % fresh culture medium addition (b), 33 °C-100 % fresh culture medium addition (c) and 33 °C-200 % fresh culture medium addition (d)
Experiment performed in spinner flask
As it was observed for experiments in cell culture plates, a stop in growth and viability drop after cell infection with SFV-GPV was also confirmed in spinner flasks (Fig. 4a). Cell death was more intensive in spinner flask than similar experiment performed in cell culture plates (Fig. 4a, 1c; Table 1). The maximum RVGP concentration was observed 8 h post-viral adsorption (MOI = 9) (Fig. 4b), 8 h earlier than that found for similar conditions in the culture plate system at MOI equal to 16 (Fig. 3c). On the other hand, volumetric yield for this glycoprotein was 2.33-fold higher in spinner flask with respect to culture plate results with equivalent experimental conditions, despite the MOI decrease (Fig. 4a, 1g). The marked decrease of RVGP concentration after maximum expression point was also confirmed in the spinner flask experiment (Fig. 4a). The RVGP mass produced per unit of virus particle in spinner flask was 0.076 pg/virus, whereas this parameter was 5.42-fold lower in culture plates.

a Viable cell concentration, cell viability and RVGP expression kinetic as well as b RVGP expression kinetic, protein production is referred per 106 cells and RVGP distribution in supernatant and cells for cell infections performed in spinner flask and MOI = 9, temperature 33 °C and 100 % fresh culture medium volume addition. Each point represent a single sample
The RVGP distribution profile was similar in spinner flask and culture plate experiments. An increase of RVGP in the extracellular environment is observed with time increase. However, the percentage of RVGP in supernatant at the end of the assessed interval was 40.8 % (Fig. 4b), while 25.3 % was detected in culture plates (Fig. 3c).
Discussion
A previous study demonstrated the practicality of BHK-21 cells to produce a recombinant RVGP at relatively high level using a recombinant SFV as expression vector. In that case, BHK-21 cells were cultured and infected with SFV-RVGPV in adherent systems (12-well culture plates) (Benmaamar et al. 2009). In the present work, BKH-21 were cultured and infected in suspension systems in order to improve the RVGP expression and to enable a future scale-up of this heterologous protein system for RVGP production. In that sense, MOI was kept constant during the block of experiments performed in cell cultures plates for allowing equivalence in experimental conditions. However, in the previous study, the initial cell concentration was 1.67 × 104 cell ml−1; it employed serum free medium and the well superficial area was twofold lower with respect to that used for the present study. A cell concentration of 2.0 × 106 cell ml−1 was utilized in suspension culture experiments. This cell population corresponds to the central point of BHK-21 exponential growth phase in suspension culture with the same culture medium, both for bioreactor (1L working volume) and spinner flask (Fernández-Núñez et al. 2014). In general, cells in good nutritional as well proper enzymatic and growth conditions, with abundant intracellular and extracellular substrate maximize virus production (Auninš 2010) and probably protein expression of “suicidal” recombinant virus like SFV-RVGPV. This justifies the choice of 48 h as time of infection.
The confirmed cell death, after SFV-RVGPV infection, is related to the cytotoxic effect on host cells caused by SFV vectors. This negative event, in conjunction with the short-term expression pattern as well as the apoptosis induction on host cells, are the major drawbacks for this transient expression system (Hassaine et al. 2006).
As a rule, the viral infection process is favored when the incubation temperature is a few degrees cooler than normal (i.e., ~33 °C instead of 37 °C). However, other studies suggest that virus is optimally produced at the higher normal temperature of 37 °C. Thus, it is recommended that a variety of conditions be examined for each virus/cell system in order to optimize virus infection (Coombs 2011). In our case, the positive impact of temperature decrease on RVGP expression was qualitatively similar to other heterologous protein expression systems using SFV vector. Increase up to 20-fold higher, has been reported when infection processes have been performed at 33 °C, instead of 37 °C (Schlaeger and Lundstrom 1998).
The effect of increase in dilution factor was not significant in the range under study (100–200 % with respect to initial cell suspension volume) on RVGP expression. In parallel, it was also confirmed that glutamine was not depleted; this nutrient is considered limiting substrate for BHK-21 growth (Fernández-Núñez et al. 2014). Thus, the increase of fresh culture medium addition after viral adsorption step would not have economic justification (unnecessary culture medium would be consumed) and downstream process could be more demanding.
The RVGP expression kinetic profiles for suspension cell culture at 37 and 33 °C (fresh culture addition 100 %) were different when compared with previous reports for adherent BHK-21 cell culture at 37 °C (Benmaamar et al. 2009). In adherent cells, the expression profile showed progressive increase over the course of monitoring time. Nevertheless, as a rule, in suspension cell, an expression peak around 16 h was observed followed by RVGP decrease. This suggests protein degradation caused by the suspension culture environment.
The RVGP expression, quantified per cell number (ng/106 cells), was similar for viral infections performed in suspension and adherent cell culture when harvest time was 16 h. However, an expression level higher than 2000 ng/106 cells was detected at the end kinetic study (48 h) for adherent cell culture (Benmaamar et al. 2009). Conversely, volumetric yield (ng ml−1) in suspension cell cultures was almost fivefold higher with respect to the value observed for this parameter in adherent cell culture. This result encourages the utilization of suspension system because of downstream process will be facilitated. On the other hand, the selection of suspension cell culture makes easier the scale-up of this bioconversion process, if necessary.
The use of fetal bovine serum in culture medium for this type of membrane glycoprotein (Ventini et al. 2010) could be avoided taking into account regulatory issues and biosafety, but it is not significant in order to improve the downstream process. The presence of this biological reagent in extracellular protein expression does have disadvantages for large-scale purification process (Bernard and Blasey 2002).
For experiment performed in cell culture plate at 33 °C, the RVGP increase in supernatant over the course of post-viral adsorption time could be associated to membrane debris caused by cell lysis after prolonged infection time with SFV vector (Benmaamar et al. 2009). This is a consequence of “shut-off” effect of the protein synthesis in host cell induced by SFV replication (Rossi et al. 2012). However, as a rule, higher percentages of RVGP were located into the cell with respect to supernatant, for all assessed experimental conditions. This is justified by the site of RVGP expression in BHK-21 cells when SFV vector is utilized. This heterologous protein is expressed on cell membrane (Benmaamar et al. 2009).
The maximum volumetric yield for RVGP (cell cultures plates with agitation) observed in the present work was lower than those reported for the same protein using stably transfected Drosophila melanogaster cells (1.5–3 mg L) with optimized culture conditions in bioreactors (Ventini et al. 2010; Rossi et al. 2012). Homogenization and aeration improvements could increase the RVGP expression. Based on this criterion, the best conditions for RVGP expression in cell culture plates were extrapolated to spinner flask.
Few papers have reported the utilization of spinner flask or bioreactor for heterologous protein expression using SFV expression system. Enzyme and cell receptors are included in this short list (Blasey et al. 2000). The reported volumetric yields are lower than 16 mg L. Probably, the present work will be one of the first papers in which spinner flask is used to obtain a heterologous viral protein with SFV vector in BHK-21 cell line.
The expression peak in spinner flask (559 ng ml−1) was higher than that observed in cell culture plates, in spite of a 56 % lower MOI with respect to the MOI used in smaller scale devices. This result demonstrated the viral infection efficiency improvement when culture conditions are enhanced. Probably, if agitation and dissolved oxygen concentration in bioreactors were studied in details, higher volumetric yield would be observed. Maybe, the relative lower protein expression yield in culture plates with respect to that observed in spinner flask could be justified by a drastic shear stress on viral vector at small scale. This event could be affect the biological activity of SFV vector and consequently, the protein expression.
At present, volumetric yield in spinner flask for RVGP are inferior to values of this parameter for the same heterologous protein and other proteins expressed with SFV vector in bioreactors (Blasey et al. 2000; Ventini et al. 2010; Rossi et al. 2012). The diminution of time for maximum protein expression peak is another evidence of improved conditions for infection in spinner flask when compared with cell culture plate. This is advantageous because the productivity is also enhanced. However, the RVGP volumetric and per cell number yields for SFV vector in combination of BHK-21 cell line cultured in suspension, both cell culture plate and spinner flask were higher than those obtained for transient expression of the same protein using plasmid vector and D. melanogaster Schneider 2 (S2) cells as a host. Specifically, in spinner flask, the RVGP per cell number yield in SFV-BHK-21 system (present study) was 40-fold higher than that reported for plasmid vector-(S2) cells (Suárez-Patiño et al. 2014). Thus, the transient SFV-BHK-21 system could be an alternative for RVGP expression for use in lab-scale (structural and functional studies).
Conclusions
Values for temperature (33 °C), fresh medium addition after viral adsorption step (100 % of fresh culture medium addition with respect to the initial cell suspension volume before infection) and protein harvest time (16 h) ensured optimal RVGP expression. The statistical tool employed for this purpose was multilevel factorial design using 24-well cell culture plates and MOI equal to 16.
The volumetric yield in these conditions was higher than those reported for adherent cell culture and S2 cells with non-viral vector. This result at lab scale could be useful for performing structural and pre-clinical studies with RVGP. In spinner flask, volumetric yield was improved, keeping constant the optimized parameters in smaller scale device and diminishing MOI (9). These results are still lower than those reported previously for the same heterologous protein with other expression systems and those with other heterologous proteins using SFV vectors, in bioreactors. Therefore, additional experiments in controlled conditions (agitation and dissolved oxygen) are suggested in order to establish a large-scale process.
References
Andréll J, Tate CG (2013) Overexpression of membrane proteins in mammalian cells for structural studies. Mol Membr Biol 30:52–63
Ansorena E, Casales E, Aranda A, Tamayo E, Garbayo E, Smerdou C, Blanco-Prieto MJ, Aymerich MS (2013) A simple and efficient method for the production of human glycosylated glial cell line-derived neurotrophic factor using a Semliki Forest virus expression system. Int J Pharm 440:19–26
Augusto EFP, Moraes AM, Piccoli RAM, Barral MF, Suazo CAT, Tonso A, Pereira CA (2010) Nomenclature and guideline to express the amount of a membrane protein synthesized in animal cells in view of bioprocess optimization and production monitoring. Biologicals 38:105–112
Auninš JG (2010) Viral vaccine production in cell culture. In: Flickinger MC (ed) Encyclopedia of industrial biotechnology: bioprocess, bioseparation, and cell technology. Wiley, New York, pp 1–52
Baldi L, Hacker DL, Adam M, Wurm FM (2007) Recombinant protein production by large-scale transient gene expression in mammalian cells: state of the art and future perspectives. Biotechnol Lett 29:677–684
Benmaamar R, Astray RM, Wagner R, Pereira CA (2009) High-level expression of rabies virus glycoprotein with the RNA-based Semliki Forest Virus expression vector. J Biotechnol 139:283–290
Bernard AR, Blasey HD (2002) Transient expression system. In: Flickinger MC, Drew SW (eds) Encyclopedia of bioprocess technology. Wiley, New York
Blasey HD, Brethon B, Hovius R, Vogel HH, Tairi AP, Lundström K, Rey L, Bernard AR (2000) Large scale transient 5-HT3 receptor production with the Semliki Forest Virus expression system. Cytotechnology 32:199–208
Cabaniols JP, Mathis L, Delenda C (2009) Targeted gene modifications in drug discovery and development. Curr Opin Pharmacol 9:657–663
Coombs KM (2011) Viruses produced from cells. In: Moo-Young M (ed) Comprehensive biotechnology, 2nd edn. Elsevier, Valencia, pp 383–393
Fernández-Núñez EG, Leme J, Parizotto LA, Chagas WA, Rezende AG, Costa BLV, Monteiro DCV, Boldorini VLL, Jorge SAC, Astray RM, Pereira CA, Caricati CP, Tonso A (2014) Influence of aeration–homogenization system in stirred tank bioreactors, dissolved oxygen concentration and pH control mode on BHK-21 cell growth and metabolism. Cytotechnology 66:605–617
Hassaine G, Wagner R, Kempf J, Cherouati N, Hassaine N, Prual C, André N, Reinhart C, Pattus F (2006) Semliki Forest virus vectors for overexpression of 101 G protein-coupled receptors in mammalian host cells. Protein Expr Purif 45:343–351
Jas D, Coupier C, Toulemonde CE, Guigal PM, Poulet H (2012) Three-year duration of immunity in cats vaccinated with a canarypox-vectored recombinant rabies virus vaccine. Vaccine 30:6991–6996
Karlsson GB, Liljestrom P (2003) Live vial vectors: Semliki Forest Virus. Methods Mol Med 87:69–82
Loza-Rubio E, Rojas-Anaya E, López J, Olivera-Flores MT, Gómez-Lim M, Tapia-Pérez G (2012) Induction of a protective immune response to rabies virus in sheep after oral immunization with transgenic maize, expressing the rabies virus glycoprotein. Vaccine 30:5551–5556
Puglia AL, Rezende AG, Jorge SA, Wagner R, Pereira CA, Astray RM (2013) Quantitative RT-PCR for titration of replication-defective recombinant Semliki Forest virus. J Virol Methods 193:647–652
Rossi N, Silva BG, Astray R, Swiech K, Pereira CA, Suazo CA (2012) Effect of hypothermic temperatures on production of rabies virus glycoprotein by recombinant Drosophila melanogaster S2 cells cultured in suspension. J Biotechnol 161:328–335
Schlaeger EJ, Lundstrom K (1998) Effect of temperature on recombinant protein expression in Semliki Forest virus infected mammalian cell lines growing in serum-free suspension cultures. Cytotechnology 28:205–211
Sevastsyanovich Y, Alfasi S, Cole J (2009) Recombinant protein production: a comparative view on host physiology. N Biotechnol 25:175–180
Suárez-Patiño SF, Mancini RA, Pereira CA, Suazo CAT, Mendonça RZ, Jorge SAC (2014) Transient expression of rabies virus glycoprotein (RVGP) in Drosophila melanogaster schneider 2 (S2) cells. J Biotechnol 192:255–262
Ventini DC, Astray RM, Lemos MAN, Jorge SAC, Calderón-Riquelme C, Torres-Suazo CA, Tonso A, Pereira CA (2010) Recombinant rabies virus glycoprotein synthesis in bioreactor by transfected Drosophila melanogaster S2 cells carrying a constitutive or an inducible promoter. J Biotechnol 146:169–172
Walsh G (2010) Post-translational modifications of protein biopharmaceuticals. Drug Discov Today 15:773–780
Zhang G, Wang H, Mahmood F, Fu ZF (2013) Rabies virus glycoprotein is an important determinant for the induction of innate immune responses and the pathogenic mechanisms. Vet Microbiol 162:601–613
Zhu J (2012) Mammalian cell protein expression for biopharmaceutical production. Biotechnol Adv 30:1158–1170
Acknowledgments
The authors would like to thank Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP: 2010/52521-6) for post-doctoral fellowship and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq: 483009/2010-5) for scientific grants. Authors are very grateful to Dr. Soraia Attie Calil Jorge, Renato Mancini Astray and Carlos Augusto Pereira for their technical suggestions and commentaries. First author gratefully acknowledges his wife and daughters, Relma, Giovanna and Paola for the inspiration to write this paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Fernández-Núñez, E.G., de Rezende, A.G., Puglia, A.L.P. et al. Transient expression of rabies virus G-glycoprotein using BHK-21 cells cultured in suspension. Biotechnol Lett 37, 1153–1163 (2015). https://doi.org/10.1007/s10529-015-1787-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-015-1787-3