
Abstract
Enterococci are ubiquitous microorganisms in almost all environments, from the soil we step on to the food we eat. They are frequently found in naturally fermented foods, contributing to ripening through protein, lipid, and sugar metabolism. On the other hand, these organisms are also leading the current antibiotic resistance crisis. In this study, we performed whole-genome sequencing and comparative genomics of an Enterococcus faecium strain isolated from an artisanal Mexican Cotija cheese, namely QD-2. We found clear genomic differences between commensal and pathogenic strains, particularly in their carbohydrate metabolic pathways, resistance to vancomycin and other antibiotics, bacteriocin production, and bacteriophage and CRISPR content. Furthermore, a bacteriocin transcription analysis performed by RT-qPCR revealed that, at the end of the log phase, besides enterocins A and X, two putative bacteriocins not reported previously are also transcribed as a bicistronic operon in E. faecium QD-2, and are expressed 1.5 times higher than enterocin A when cultured in MRS broth.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Members of the Enterococcus genus are non-motile, non-sporulated, Gram-positive lactic acid bacteria (LAB) distributed ubiquitously in many environments. They commonly live in water, soil, animal gut microbiome, and even human milk; they also belong to dairy farm environments such as feed, bedding, and udder and teet surfaces. Consequently, these microorganisms can also be found in food derived from the above sources, e.g., fermented meats and vegetables and dairy derivatives such as cheese (Ben Braïek and Smaoui 2019). It is estimated that enterococci can make up to 1% of the total microbiome of handmade artisanal cheeses, similar to the human fecal microbiota composition (Dubin and Pamer 2017; Escobar-Zepeda et al. 2016).
Enterococci, however, are also opportunistic pathogens in certain niches, namely hospital environments, because some strains contain a variety of virulence factors and are intrinsically resistant to many common antibiotics such as β-lactams, aminoglycosides, and macrolides (Freitas et al. 2021). Particularly, certain Enterococcus faecium strains are resistant to vancomycin (vancomycin-resistant enterococci, VRE)—a phenotype commonly associated with inpatient enterococcal infections—and are of concern worldwide, being at the top of the WHO list of priority pathogens. Currently, VRE are a major target in the development of new antibiotics (World Health Organization 2017). Also, enterococci are a natural reservoir of multiple virulence factors and antibiotic-resistance genes. Unlike most LAB, the genus is neither considered Generally Regarded As Safe (GRAS) nor does it have the Qualified Presumption of Safety (QPS) status, and its role as a probiotic or starter culture is still debated (Dapkevicius et al. 2021).
Virulence factors have distinct roles during the infection process. While some elements in E. faecium pathogenic strains may facilitate host invasion (cytolysin, hyaluronidase, gelatinase), other factors participate only in the colonization stage (i.e., aggregation substance, collagen-binding protein, surface protein) (Chajęcka-Wierzchowska et al. 2017). Both pathogenic and commensal enterococcal strains require colonization factors, but only the pathogenic ones contain the invasion elements needed to harm the host (Dapkevicius et al. 2021). Even if commensal strains contain genes related to antibiotic resistance, these are useless because commensal strains lack the invasiveness needed to spread within the host (Beceiro et al. 2013).
In artisanal cheeses made from raw milk, E. faecium plays a role in the ripening process via the synthesis of various substances related to the development of textures, odours, and flavours. It also produces various antimicrobial substances that contribute to the innocuity of cheese and preserves it with no need for processes such as pasteurization (Olvera-García et al. 2018). Additionally, commensal strains possess certain genotypical and phenotypical characteristics that allow them to act as probiotics through bowel colonization, which depends on factors such as the aggregation substance and the surface protein (Chajęcka-Wierzchowska et al. 2017). Probiotics may benefit the host by enhancing nutrient and drug bioavailability, regulating the immune system, and boosting the neurological system (Maldonado Galdeano et al. 2019). It is clear that the behaviour of commensal strains differs substantially from the one of their pathogenic relatives.
Given the considerable amount of genotypical and phenotypical variations within the same species, genome-wide studies are an effective method to determine which elements make a strain pathogenic or commensal. In these studies, researchers look for genetic features related to invasion, colonization, antibiotic resistance, immune system evasion, toxin production, biomolecule uptake and usage, flavour and odour production, and bacteriocin synthesis, among others (Apostolakos et al. 2023). The present study explored the genome of E. faecium QD-2 strain isolated from an artisanal Cotija cheese prepared locally in the Jalisco-Michoacán region of Mexico via genome-wide studies and RT-qPCR of bacteriocin transcripts.
Materials and methods
Strain isolation and DNA extraction
LAB isolation was performed from seven “Region of Origin” artisanal Cotija cheeses. After 10 g of each cheese was homogenized using a Stomacher 400 Circulator (Seward Laboratory, London, UK), with 90 mL of peptone water (0.85% NaCl, w/v; casein peptone 1%, w/v), an enrichment step in Man, Rogosa & Sharpe broth (MRS, Oxoid, Basingstoke, UK) was carried out (37 °C, 250 rpm, 24 h). An aliquot of this culture was inoculated in the selective EVA (Ethyl-Violet-Azide) medium (Condalab, Madrid, Spain) (37 °C, 250 rpm, 48 h) and later streaked in Kanamycin-Aesculine-sodium Azide (KAA, Oxoid) agar. Thirty-nine colonies with the characteristic Enterococcus phenotype were selected (kanamycin resistance and black halos due to the hydrolysis of aesculine). Out of these, the 12 isolates which showed a 1090 base pairs (bp) amplicon corresponding the ddl (D-Ala:D-Ala ligase) gene, using the primers reported by Depardieu et al., (2004), corresponded to Enterococcus faecium. These strains also showed a catalase ( +) and Gram ( +) phenotype. Finally, the antilisterial activity of all strains was tested by agar diffusion tests, in which strain QD-2 showed the largest zone of inhibition. E. faecium QD-2 (CFQ-B-304) was deposited in the Culture Collection of the School of Chemistry (CFQ) at Universidad Nacional Autónoma de México (UNAM) (WDCM No. 100). For DNA extraction, QD-2 was subcultured statically in MRS broth (Oxoid) at 37 °C for 48 h. DNA was extracted as described by Sambrook et al. (2012).
DNA sequencing and assembly
Total DNA was sequenced in an Illumina NextSeq 500 platform at the sequencing unit of the Unidad Universitaria de Secuenciación Masiva y Bioinformática-UNAM (Massive Sequencing and Bioinformatics University Unit; UUSMB-IBt-UNAM) in a paired-end format, with a depth of 5,000,000 reads and a read length of 75 bp. Read quality and GC content were assessed using the FastQC software (Andrews 2010). Reads were assembled using Spades v3.13.3 (Prjibelski et al. 2020). Assembly integrity and contamination were estimated using CheckM v1.0.18 in KBase (Parks et al. 2015). The scaffolds were oriented in Mauve v20150226 using E. faecium ATCC 8459 (GCA_000336405.1) as a template (Darling et al. 2004). N50 and L50 statistics were calculated using an in-house Perl script. Coding regions were predicted and annotated using Prokka v1.13.3 (Seemann 2014) and Parallel Annotation Pipeline (PAP) v1.0 (Estrada 2023). The dataset generated during the present study is available on GenBank under accession number CP130822.
Genomic analyses
Five pathogenic (Aus0004, Aus0085, DO, V1164 and V1836; GCA_000250945.1, GCA_000444405.1, GCA_000174395.2, GCA_020162175.1 and GCA_008728455.1, respectively) and four dairy-related E. faecium strains (D, ATCC 8459, DRD-156 and IQ110; GCA_002006745.1, GCA_000336405.1, GCA_023743905.1 and GCA_001455445.1, respectively) were selected to carry out the comparative genomic analyses against QD-2. Pangenomic clustering of all ten strains was performed using the Cluster of Orthologous Groups (COG) and OrthoMCL algorithms, and a maximum likelihood tree was generated using GET_HOMOLOGUES v3.5.5 (Contreras-Moreira and Vinuesa 2013). The pangenomic structure was graphically represented using Roary v3.11.2 (Page et al. 2015). The genomic contents of all strains was further analysed using OrthoVenn v3.0, using the OrthoFinder algorithm (Sun et al. 2023).
Virulence factors were surveyed in the Virulence Factor Database (VFDB) (Chen 2004), and antibiotic resistance genes were screened using AMR++ v3.0 (Bonin et al. 2022). Genomes were screened for CRISPR-Cas sequences using CRISPRCasFinder (Couvin et al. 2018), and bacteriophage contents were explored using PHASTER (Arndt et al. 2016). Integron presence was assessed using Integron_Finder v2.0.2 (Néron et al. 2022).
The presence of genes related to sugar metabolism was confirmed using dbCAN3 (Zheng et al. 2023), and genes related to proteolysis, lipolysis, and biogenic amine synthesis were detected using the KEGG Automatic Annotation Server (KAAS) (Moriya et al. 2007).
A prediction of bacteriocin-coding sequences in E. faecium QD-2 was carried out using BAGEL4 v1.2 (van Heel et al. 2018), and the resulting sequence identities were confirmed using the blastp suite in NCBI and UNIPROT (Altschul et al. 1990). Sigma 70 promoter sequences were predicted using IPro70-FMWin (Rahman et al. 2018).
Bacteriocin expression
E. faecium QD-2 was cultured and incubated statically in 25 mL MRS broth (Oxoid) at 37 °C for 18 h. Its growth curve was plotted, and the optimal timepoint for RNA extraction was determined as six hours (end of the log phase). RNA was extracted with the TRIzol method (TRI Reagent®, Sigma-Aldrich, St Louis, MO) as described by the manufacturer. RNA integrity was assessed by agarose gel electrophoresis (1% agarose–18% formaldehyde, Sigma-Aldrich). The transcripts of interest were amplified via RT-qPCR using the SCRIPT RT-qPCR SybrMaster kit (Jena Bioscience, Dortmund, Germany), following the manufacturer’s instructions. Primers were designed from the predicted bacteriocin-coding open reading frames (ORFs) and synthesised by Integrated DNA Technologies, Inc. (Coralville, IA) (Table 1).
The relative expression of the putative bacteriocins of interest (ORFs 615, 616, 901, and 902) against the reference gene (enterocin A, ORF 621) was calculated using the ΔΔCt method. It consists of the normalization of the respective cycle threshold (Ct) values against an endogenous reference gene (in our case, rpoA) and the comparison of the normalised expression (ΔCt) against a reference gene of interest (ΔΔCt), expressed as 2−ΔΔCt (Livak and Schmittgen 2001).
Statistical analysis
RT-qPCR data were subjected to a one-way Analysis of Variance (ANOVA) (α = 0.05) in R v4.2.3 and GraphPad Prism v8. The normality of the dataset was assessed using Q-Q plots and the Shapiro–Wilk test. The data homoscedasticity was determined using Bartlett’s test, and post-hoc differences between groups were calculated using Tukey’s range test.
Results and discussion
The present study was carried out to assess the similarities and differences between a food Enterococcus faecium strain (QD-2) isolated from a naturally ripened Mexican cheese (Cotija cheese) against four other dairy-related strains (D, ATCC 8459, DRD-156 and IQ110) and five pathogenic ones (Aus0004, Aus0085, DO, V1164 and V1836). Strain D was also isolated from Cotija cheese (Olvera-García et al. 2018).
Comparative genomics
Total DNA extraction from E. faecium QD-2 revealed the absence of plasmid material; the FastQC read quality evaluation at each nucleotide position was ≥ 26, which is considered adequate. Table 2 shows the statistical analyses of the sequencing procedure. Since integrity was greater than 99% and contamination was 0%, the draft genome was considered suitable for the subsequent bioinformatic analyses.
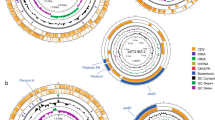
First, the pangenomic structure of the ten strains was determined, showing a clear division into two distinct clades (pathogenic and commensal) (Fig. 1). The genomic core contains all genetic elements that are essential for living and those that characterise the Enterococcus genus. Of particular interest are the features that vary between pathogenic and commensal strains. The genomic plasticity of the species can be visually appreciated by simply comparing the absence or presence of certain genomic elements between strains. These include mobile elements (phage-related), virulence factors, antibiotic resistance, metabolic features, and bacteriocin-coding genes, among others (Fig. 1 and Table 3, 4, 5 and S1). Studies on E. faecium, E. faecalis, and E. hirae have shown that, even at the species level, virulence-associated elements, such as biofilm formation and cell adhesion, and carbohydrate metabolism are highly niche-dependent (Zaidi et al. 2022). Additionally, food-related enterococcal strains tend to have multiple copies of those genes required to use specific carbohydrates, such as lactose, and of certain proteolytic genes due to the selective pressure of the medium (Olvera-García et al. 2018). Enterococci possess high genomic flexibility, allowing them to acquire various mobile elements associated with pathogenicity and survival capability in a wide range of ecosystems (Boumasmoud et al. 2022). However, the acquisition and usage of these elements are highly dependent on the niche and the conditions in which the microorganism inhabits (Das et al. 2022).

Pangenomic structure and maximum likelihood tree of Enterococcus faecium strains: five pathogenic (Aus0004, Aus0085, DO, V1164 and V1836) and five commensal strains (ATCC 8459, D, QD-2, DRD-156 and IQ110)
Venn diagrams are useful for comparing the shared elements between organisms. In the present study, these diagrams reveal that their pangenome comprise 3355 genes, out of which 2082 belong to the core genome. Accessory genes shared between QD-2 and the pathogenic strains comprise around 5.5% of the pangenome, i.e. 184 genes (Fig. 2). A manual parsing of these sequences indicates that most correspond to basic metabolic processes such as carbohydrate intake (40%, 73 genes) and bacteriophage-related proteins (20%, 37 genes). This figure differs from the 462 accessory genes shared exclusively between all pathogenic strains, some of which encode proteins related to antibiotic resistance (vanWSB, tetR) and plasmid mobilization (mobC, traE, and ardA), the latter encoding an anti-restriction protein that facilitates the establishment of mobile elements in the microorganism and it is present in multiple copies in the pathogenic strains: up to three in Aus0004, Aus0085, V1164 and V1836. Anti-restriction proteins have been associated with antibiotic resistance dissemination, such as carbapenem resistance in Klebsiella pneumoniae because they prevent foreign DNA restriction (Liang et al. 2017). The presence of these DNA mobilization elements in the pathogenic strains is consistent with their genomic structure, as all possess multiple plasmids (between 3 and 9); whereas the commensal strains D and QD-2 have none and strains ATCC 8459 and DRD-156 have only one.

Core-, pan- and unique genome structure of all food (ATCC 8459, D, DRD-156, IQ110 and QD-2) and pathogenic (Aus0004, Aus0085, DO, V1164 and V1836) strains
Bacteriophage infection remnants
The analysis of the ten genomes using PHASTER revealed multiple regions with bacteriophage identity. These elements were compared in Table 3, which shows only those sequences with a completeness score higher than 100, calculated using the platform as described by Arndt et al. 2016. Bacteriophages can make up to 20% of the total genomic content of bacteria and can provide the metabolic means needed to adapt to a particular niche (Casjens 2003). Our findings show that, compared to the pathogenic strains, commensal Enterococcus contain fewer prophage sequences inserted into their genomes, and the predominant ones come from LAB-infecting bacteriophages, such as Lactobacillus and Enterococcus phages. On the other hand, pathogenic strains contain more Listeria, Staphylococcus and Enterococcus prophages. This pattern suggests the infection history of those strains and the kind of interactions of certain E. faecium strains with their environment. This bacteriophage-mediated horizontal transfer behaviour within and between phyla plays a central role in bacterial evolution, and these interactions may trigger the transfer of certain genetic elements pertaining to pathogenicity and virulence in a specific niche, such as a hospital setting (Casjens and Hendrix 2014; Ikhimiukor et al. 2023). It has been reported that certain Gram-negative-infecting viral families, such as Inoviridae, transport genes related to biofilm synthesis, immune evasion, and toxin secretion (Burckhardt et al. 2023). Thus, it is not surprising that specific Enterococcus strains may have acquired some of their genetic elements from this cross-genus horizontal gene transfer, which is highly dependent on their environment.
Likewise, this behaviour depicts the receptiveness of pathogenic E. faecium strains in regard to exogenous DNA. Bacteria defend themselves from bacteriophage infections by means of systems such as CRISPR-Cas, a bacterial immune system that functions through the insertion of small viral fragments into the bacterial genome and the subsequent recognition and restriction of these sequences in case of a second infection. Therefore, this system plays a major role in the evolution of bacterial genomes (Rath et al. 2015). It was observed that none of the studied strains contains a functional CRISPR-Cas system; all contain CRISPR sequences, but no Cas coding genes inserted in their genomes. It has been described that multidrug-resistant E. faecium strains lack a functional CRISPR-Cas system (Alduhaidhawi et al. 2022). This may imply that, in a hospital setting, these strains are subjected to a selective pressure favouring the survival of strains unable to decompose exogenous DNA; consequently, they can easily acquire mobile virulence and resistance genes, which may help them thrive in this environment (Tao et al. 2022). This behaviour has been observed and described in VRE E. faecium and E. faecalis (Alduhaidhawi et al. 2022). On the other hand, food strains are not subjected to this pressure; as a result, there is no clear preference towards the predominance of either CRISPR-Cas-competent or incompetent organisms (Markusková et al. 2018; Oliveira et al. 2022). This study determined that none of the ten strains contain integrons in their genome, but some contain multiple integrase-coding genes. As already mentioned, bacteriophage sequences in the bacterial genome are associated with virulence (Casjens 2003). Particularly, class-1 integrons are responsible for the spread of antibiotic resistance in most Gram-negative pathogenic bacteria and a few Gram-positive ones (Ghaly et al. 2017).
Virulence attributes and antibiotic resistance elements
It should also be noted that both commensal and pathogenic strains share multiple biofilm (bopD), capsule (cpsA, cpsB), and adhesion (efaA, acm, ebp) virulence factor genes according to VFDB, which are necessary for their establishment in the host (Table 4). These properties are desirable in potentially probiotic Enterococcus strains, and their presence in the probiotic strain E. faecalis Symbioflor1 has been reported (Domann et al. 2007).
Despite sharing multiple virulence factors, none of the commensal strains contains the enterococcal surface protein gene, esp. Enterococcal strains containing esp usually also carry vancomycin-resistance genes (Kafil and Mobarez 2015). Additionally, a manual parsing of the annotated genomes of the pathogenic strains reveals the presence of hlyA (haemolysin A), hlyIII (haemolysin III), and hlyC (the haemolysin transport protein). These are virulence factors related to the invasiveness and toxicity of the bacteria, and are absent in the genomes of the commensal strains (Chajęcka-Wierzchowska et al. 2017).
None of the commensal strains harbour vancomycin-resistance genes; by contrast, most pathogenic strains have five, thus belonging to the VRE group and, consequently, being of high epidemiologic relevance (Centers for Disease Control and Prevention 2019). The development of multidrug-resistant strains from those initially innocuous has been exacerbated in the past 50 years, and this behaviour is strongly related to human activity (Beceiro et al. 2013). Some of the commensal strains studied contain the innate betalactam, aminoglycoside and macrolide resistances expected in the genus (Table 5) (Freitas et al. 2021) and they also harbour resistance genes to less common antibiotics such as fluoroquinolones and elfamycins. To successfully deal with antibiotics, not only resistance genes are important, but also antibiotic efflux pumps. Genes encoding these membrane proteins were found in all ten strains; however, it is remarkable that Aus0085 is not only resistant to a broader range of antibiotic categories (nucleosides and tetracyclines) but also contains additional efflux pumps that are absent in their counterparts. The inherent antibiotic resistance may result from natural selective pressure to adapt to a particular niche and does not necessarily reflect human activity. Enterococcus does not exclusively inhabit the animal gut but is also found in plants, sand, and soil (Dapkevicius et al. 2021). These habitats also harbour Streptomyces, an actinobacterium from which many widely used antibiotics are produced, such as kanamycin, streptomycin, erythromycin, tetracyclin, and even vancomycin (Quinn et al. 2020). This microorganism uses these molecules as defence mechanisms and has developed intrinsic resistance to them, which can be transferred to other microorganisms via horizontal gene transfer (Seipke et al. 2012). Thus, a primitive Enterococcus strain may have acquired these resistance genes by interacting with Streptomyces; over time, the strains that acquired these genes may have been favoured by the environment to survive, creating the intrinsically resistant genus we know today. The pool of antibiotic resistance genes in the environment that can be horizontally transferred between organisms is called the environmental resistome and is distributed in both commensal and pathogenic microorganisms (Wright 2010). Furthermore, it is not just bacteria that contribute to the environmental resistome, as mobile resistance genes also occur in environmental bacteriophages, making them part of this phenomenon (Das et al. 2022). To note, this study found that the plasmids belonging to Aus0085, DO, V1164 and V1836 contain antibiotic resistance genes (lsa, ermB, aph-D, ant(6’), lsaE, sat, catA), while the plasmids from commensal strains do not possess any resistance genes. It is worth mentioning that strains V1164 and V1836 harbour all of their vancomycin resistance genes in their plasmids, while Aus0004 and Aus0085 carry them in their chromosome.
Enterococcal metabolic features relevant to food fermentation and preservation
On the other hand, QD-2 shares 248 accessory elements with the other commensal strains. Of these, those related to carbohydrate metabolism constitute the predominant category, with transport-system genes for PTS sugars, such as mannitol, ascorbate, sorbitol, and glucitol/sorbitol. PTS is a phosphotransferase system that facilitates the metabolism of many monosaccharides, disaccharides, and other carbohydrate derivatives that depend on phosphoenolpyruvate. For microorganisms growing in dairy products such as cheese, possessing the necessary means to internalise sugars using PTS is of utmost importance (Flórez and Mayo 2015). In dairy products, lactose is the main carbon source for Enterococcus; however, this disaccharide is not the only carbohydrate that this microorganism can use. This was demonstrated with the dbCAN3 tool, which found several starch, sucrose, xylan, α/β-glycan, trehalose, pectin, chitin, rhamnose, and arabinan hydrolases in the QD-2 genome. Chitin is the second most abundant polysaccharide in nature, after cellulose. The ability to degrade cellulose has been previously described in gut enterococcal strains, and their chitin-degrading capacity has been shown to inhibit the growth of Fusarium solani, a phytopathogenic fungus (Atwa et al. 2022; Kohl et al. 2022). In the context of the artisanal Cotija cheese, E. faecium QD-2 might be capable of using yeast debris as a carbon source, as these are part of the cheese microbial community (Escobar-Zepeda et al. 2016).
Another set of important elements associated with cheese ripening found in the commensal strains is the proteolytic system. LAB are microorganisms that require an external source of certain amino acids, as they are auxotrophic to many of them (Dapkevicius et al. 2021). To meet their amino acid requirements, these dairy microorganisms break down and metabolise casein from the medium to grow and develop (Martino et al. 2018). This is useful not only for these microorganisms but also for the manufacturers of fermented food products, as proteolysis is one of the main sources of flavours and textures in fermented food (Dapkevicius et al. 2021). Previously, we characterised the proteolysis system in E. faecium D, it contains a large variety of genes encoding proteins that break down (Clp), internalise (Opp, Dpp, Dpt), and further metabolise casein from the medium (Pep) (Olvera-García et al. 2018). In the present study, we observed that strain QD-2 possesses almost all the genes encoding the proteins previously described, in addition to many components absent in strain D, such as clpQ, clpC, and pepP (Table S1). The generation of bitter flavours could be controlled by PepP, an aminopeptidase that tightly controls the production and breaking down of Xaa-Pro-Xaa peptides, which are responsible for producing bitter flavours in cheese (Savijoki et al. 2006).
Another flavour-related metabolic feature found in food-enterococcal strains is the lipolytic system, producing compounds such as esters and free fatty acids, which give fermented foods their characteristic flavour and aroma (Martino et al. 2018). Many compounds derived from lipolysis and fatty acid metabolism, such as 2-heptanone, 2-nonanone, butanoic, hexanoic, and octanoic acids, are fragrant (Vélez et al. 2023). Our previous study showed that E. faecium D contains genes related to lipolysis, such as a lipase, two acetylesterases, and a carboxylesterase (Olvera-García et al. 2018). QD-2 genome annotation through KAAS reveals the presence of genes encoding one tributyrin esterase (estA), two acetylesterases (aes), and two carboxylesterases (yvaK, QD2_01579). It has been described that tributyrin esterase, EstA, is a key protein in the development of desirable aromas in cheese, alongside the aminopeptidase PepP previously described (Engels et al. 2022).
Other aroma-associated molecules produced by fermentation are volatile compounds such as ethanol, acetate, acetone, diacetyl, acetoin, and 2, 3-butanediol (Sarantinopoulos et al. 2002; Martino et al. 2018). Analysis of the QD-2 genome unveils the presence of genes encoding enzymes related to the production of acetoin (ilvBIG, alsCD, budA), ethanol, acetaldehyde, and acetate (adhEC). Furthermore, it was also found that QD-2 possesses the citrate operon citCDEFGX. Many aromas found in cheese, such as acetate, may also be derived from citrate metabolism (Sarantinopoulos et al. 2002).
Besides desirable compounds, some molecules produced during food fermentation are biogenic amines, such as putrescin, tyramine, histamine, and cadaverine, which are products of the decarboxylation process of ornithine, tyrosine, histidine, and lysin, respectively (Olvera-García et al. 2018). The undesirable presence of these compounds in food is commonly associated with health disorders for the consumer, such as diarrhoea, headache, vomiting, and tachycardia, and could also have a direct effect on carcinogenesis. The presence of biogenic amines is usually an indicator of food quality (Wójcik et al. 2020). We previously showed that although E. faecium D contains the genes necessary to produce tyramine, putrescin, and ornithine, these compounds could not be detected at significant levels in Cotija cheese via HPLC (Olvera-García et al. 2018). The analysis of the E. faecium QD-2 genome reveals the presence of tyrCD, aguA, and argF, associated with the synthesis of the biogenic amines mentioned above. The presence of these genes in the genome of QD-2 may not correlate with the significant production of biogenic amines in the same way as in strain D.
Bacteriocin-associated elements were shared between the commensal strains, corresponding to enterocin B (entB), a bacteriocin-immunity protein, and ABC transporters, except strain IQ110 which lacks entB. Enterocin B found in strains D, QD-2 and ATCC 8459 was identical, sharing 78% identity with enterocin B from QD-2 and 53% with carnobacteriocin A from Carnobacterium maltaromaticum. Enterocin B is an antimicrobial peptide belonging to the IIa bacteriocin group that possesses substantial activity against many food-related pathogens, i.e., Listeria monocytogenes (Ankaiah et al. 2018). The evolutionary history of Enterococcus suggests that the genus emerged as a ramification of Carnobacterium, which, in turn, evolved from a Vagococcus-like ancestor (Dapkevicius et al. 2021). An ancestral carnobacteriocin A-like molecule was probably encoded in the common ancestor of Enterococcus and Carnobacterium; later, as evolution took place, this primitive bacteriocin diverged into enterocin B and carnobacteriocin A since both are very similar (Casaus et al. 1997). This divergence still prevails, as certain variants of enterocin B only share 60% identity with other enterocin B variants (Qiao et al. 2020).
Bacteriocin expression in Enterococcus faecium QD-2
Enterococcus is one of the most diverse genera in bacteriocin production, as these peptides are active against a wide variety of phyla, being of special interest given the current antibiotic resistance crisis (Almeida-Santos et al. 2021). Based on the whole-genome sequencing of QD-2 and its analysis on BAGEL4, eight bacteriocins were predicted in its genome, whose ORFs correspond to 615 and 616 (predicted bacteriocins), 621 (enterocin A, entA, Wu et al. 2022), 718 (uviB-like, Dupuy et al. 2004), 901 and 902 (enterocin X, enxA/enxB, Hu et al. 2010), 2142 (enterolysin A, Nilsen et al. 2003), and 2596 (enterocin B, entB, Casaus et al. 1997). It is worth noting that the activity of all but three (ORFs 615, 616, and 718) have been previously characterised. However, sequences 615 and 616 were identified as putative bacteriocins by O’Keeffe et al. (1999) (herein ORF1 and ORF2, respectively). More recently, our team identified enterocin 29 (Ent29α), a bacteriocin produced by an enterococcal strain found in a fermented sausage, which shares 100% identity with ORF 616 after its signal peptide has been excised and contains a consensus sequence related to antilisterial activity (Escamilla-Martínez et al. 2017). The putative bacteriocins 615 and 616 contain the signal peptide cleavage sequence GG, present in class II bacteriocins (Nes et al. 2013).
Enterocin A is particularly relevant for being the first widely studied class-IIa bacteriocin of enterococcal origin, especially for its activity against the foodborne pathogen L. monocytogenes (Wu et al. 2022). Enterocin B is a bacteriocin with activity against many pathogens such as Acinetobacter baumanii, Staphylococcus aureus, and L. monocytogenes; it also inhibits the formation of biofilms and possesses activity against human cancer cell lines (Ankaiah et al. 2018; Wu et al. 2022). Additionally, enterocin B and enterocin A can act synergically with one another, enhancing their antimicrobial activity (Casaus et al. 1997). Similarly, enterocin X is a class-IIb bacteriocin formed by two synergic peptides (Xα and Χβ) and can be found in E. faecium strains that produce enterocin A and B. It has been reported that ORFs 901 and 902 share the GXXXG motifs usually found in enterocin X (Hu et al. 2010).
E. faecium QD-2 has shown antilisterial activity in agar diffusion tests. Based on the available information about the biochemical characterization of enterocins A and X (ORFs 621, 901, and 902, respectively), the present study focused on ORFs 615 and 616 (Mendoza 2019). We assessed the transcription probability of these ORFs through an in-silico analysis of their promoter sequences and contrasted it against ORFs 901 and 902. iPro70-FMWin finds patterns by comparing canonical sigma 70 promoter sequences, considers the distance between regions -10 and -35, and calculates the probability of a functional promoter; thus, a score close to 1 represents a highly probable promoter sequence (Rahman et al. 2018). Two highly probable promoter sequences were located upstream from ORFs 615 and 901, with scores of 0.7860/0.7860 and 0.9981/0.9981 for regions -10/-35, respectively, while ORFs 616 and 902 showed lower scores, 0.0914/0.0914 and 0.4973/0.1140, respectively. These results suggest that these ORFs could be organised as bicistronic operons, which makes sense for enterocin X, described as a class-IIb two-peptide bacteriocin (Wu et al. 2022).
The transcription rate of ORFs 615, 616, 901, and 902 was evaluated through an RT-qPCR. The housekeeping gene rpoA, which encodes the α-subunit of RNA polymerase, was selected as the normalization gene because its expression is fairly constant. It has proven reliable in RT-qPCR procedures in Clostridium perfringens and Campylobacter jejuni (Williams and Ghanem 2022; Ritz et al. 2009). The expression of ORF 621 was also considered as a baseline. All Ct values obtained from our analysis yielded a variance ratio lower than 1%, suggesting that the experimental procedure was reproducible and the results are reliable (Fig. 3).

Ct values for all transcripts. Different letters represent a significant difference. (\(\alpha\)< 0.05)
Normalised ΔCt values of ORFs 615, 616, 901, and 902 were compared against the ΔCt value for enterocin A (ORF 621) to calculate their relative expression, ΔΔCt. (Fig. 4). A clear difference between the expression of 615–616 and 901–902 relative to 621 can be observed, but there is no significant difference between the expression of 615 and 616, nor between 901 and 902. A one-way ANOVA and a post-hoc Tukey test found significant differences between all genes, except within 615–616 (p = 0.9995) and 901–902 (p = 0.9999). This behaviour, coupled with the strength of the promoters found upstream of these ORFs, suggests that putative bacteriocins 615–616 and 901–902 are co-expressed. This is also consistent with the genomic proximity between them. The expression of operons 615–616 and 901–902 is 1.5 times higher and 0.8 times lower, respectively, compared to enterocin A.

Relative expression of ORFs 615, 616, 901, and 902 compared to 621 (dotted line). Different groups represent a significant difference (NS = p > 0.05, *** = p < 0.05)
Conclusions
The current discussion on the probiotic and biotechnological applications of enterococci, as well as on clinical concerns, is still a widely debated topic. We have observed that enterococci harbour many virulence and resistance factors in their genome, whether from hospital or food-related environments. However, the specific factors contained in and expressed by a given strain are highly dependent on the environment, as not all niches share the same stress conditions. Pathogenic strains contain more genes related to multidrug resistance, colonization ability, and host toxicity, whereas food-related enterococcal strains tend to carry genes related to flavour, aroma, texture development, carbohydrate uptake, lipolysis, and bacteriocins. Particularly, the E. faecium QD-2 strain expresses bacteriocins A and X, in addition to two putative additional ones encoded by ORFs 615 and 616, which need to be further characterised. The difference between strains from different environments is clear evidence of the influence of human activities on these organisms, as they are essential components not only of our own microbiota but also of the food we eat and the places we inhabit.
Data availability
The datasets generated during and/or analysed during the current study are available in the Genbank repository under accession number CP130822.
References
Alduhaidhawi AHM, AlHuchaimi SN, Al- Mayah TA, Snr A-O, Alkafaas SS, Muthupandian S, Saki M (2022) Prevalence of CRISPR-Cas systems and their possible association with antibiotic resistance in Enterococcus faecalis and Enterococcus faecium collected from hospital wastewater. IDR 15:1143–1154. https://doi.org/10.2147/IDR.S358248
Almeida-Santos AC, Novais C, Peixe L, Freitas AR (2021) Enterococcus spp. as a producer and target of bacteriocins: a double-edged sword in the antimicrobial resistance crisis context. Antibiot 10:1215. https://doi.org/10.3390/antibiotics10101215
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410. https://doi.org/10.1016/S0022-2836(05)80360-2
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc. Accessed 5 July 2023
Ankaiah D, Palanichamy E, Antonyraj CB, Ayyanna R, Perumal V, Ahamed SIB, Arul V (2018) Cloning, overexpression, purification of bacteriocin enterocin-B and structural analysis, interaction determination of enterocin-A, B against pathogenic bacteria and human cancer cells. Int J Biol Macromol 116:502–512. https://doi.org/10.1016/j.ijbiomac.2018.05.002
Apostolakos I, Paramithiotis S, Mataragas M (2023) comparative genomic analysis reveals the functional traits and safety status of lactic acid bacteria retrieved from artisanal cheeses and raw sheep milk. Foods 12:599. https://doi.org/10.3390/foods12030599
Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS (2016) PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res 44:W16–W21. https://doi.org/10.1093/nar/gkw387
Atwa N, Wahba M, Maany D, Awad H, Abo-Alkasem M, El-Masry H, Amer M, El-Diwany A (2022) Lactic acid bacteria: economic propagation, chitinases activity, and enhancing viability by gel encapsulation. Egypt Pharmaceut J 21:347. https://doi.org/10.4103/epj.epj_50_22
Beceiro A, Tomás M, Bou G (2013) Antimicrobial resistance and virulence: A successful or deleterious association in the bacterial world? Clin Microbiol Rev 26:185–230. https://doi.org/10.1128/CMR.00059-12
Ben Braïek O, Smaoui S (2019) Enterococci: between emerging pathogens and potential probiotics. BioMed Res Int 2019:1–13. https://doi.org/10.1155/2019/5938210
Bonin N, Doster E, Worley H, Pinnell LJ, Bravo JE, Ferm P, Marini S, Prosperi M, Noyes N, Morley PS, Boucher C (2022) MEGARes and AMR++, v3.0: an updated comprehensive database of antimicrobial resistance determinants and an improved software pipeline for classification using high-throughput sequencing. Nucleic Acids Res. https://doi.org/10.1093/nar/gkac1047
Boumasmoud M, Denglerhaunreiter V, Schweizer TA, Meyer L, Chakrakodi B, Schreiber PW, Seidl K, Kühnert D, Kouyos RD, Zinkernagel AS (2022) Genomic surveillance of vancomycin-resistant Enterococcus faecium reveals spread of a linear plasmid conferring a nutrient utilization advantage. Mbio. https://doi.org/10.1128/mbio.03771-21
Burckhardt JC, Chong DHY, Pett N, Tropini C (2023) Gut commensal Enterocloster species host inoviruses that are secreted in vitro and in vivo. Microbiome. https://doi.org/10.1186/s40168-023-01496-z
Casaus P, Nilsen T, Cintas LM, Nes IF, Hernández PE, Holo H (1997) Enterocin B, a new bacteriocin from Enterococcus faecium T136 which can act synergistically with enterocin A. Microbiol 143:2287–2294. https://doi.org/10.1099/00221287-143-7-2287
Casjens S (2003) Prophages and bacterial genomics: What have we learned so far? Mol Microbiol 49:277–300. https://doi.org/10.1046/j.1365-2958.2003.03580.x
Casjens S, Hendrix RW (2014) Bacteriophages and the bacterial genome. Bact Chromosome. https://doi.org/10.1128/9781555817640.ch3
Centers for Disease Control and Prevention (2019) Vancomycin-resistant Enterococci (VRE) in Healthcare Settings. https://www.cdc.gov/hai/organisms/vre/vre.html. Accessed 5 July 2023
Chajęcka-Wierzchowska W, Zadernowska A, Łaniewska-Trokenheim Ł (2017) Virulence factors of Enterococcus spp. presented in food. LWT 75:670–676. https://doi.org/10.1016/j.lwt.2016.10.026
Chen L (2004) VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res 33:D325–D328. https://doi.org/10.1093/nar/gki008
Contreras-Moreira B, Vinuesa P (2013) GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl Environ Microbiol 79:7696–7701. https://doi.org/10.1128/aem.02411-13
Couvin D, Bernheim A, Toffano-Nioche C, Touchon M, Michalik J, Néron B, Rocha EPC, Vergnaud G, Gautheret D, Pourcel C (2018) CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res 46:W246–W251. https://doi.org/10.1093/nar/gky425
Darling ACE, Mau B, Blattner FR, Perna NT (2004) Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14:1394–1403. https://doi.org/10.1101/gr.2289704
Das S, Bombaywala S, Srivastava S, Kapley A, Dhodapkar R, Dafale NA (2022) Genome plasticity as a paradigm of antibiotic resistance spread in ESKAPE pathogens. Environ Sci Pollut Res 29:40507–40519. https://doi.org/10.1007/s11356-022-19840-5
Dapkevicius MLE, Sgardioli B, Câmara SPA, Poeta P, Malcata FX (2021) Current trends of enterococci in dairy products: a comprehensive review of their multiple roles. Foods 10:821. https://doi.org/10.3390/foods10040821
Domann E, Hain T, Ghai R, Billion A, Kuenne C, Zimmermann K, Chakraborty T (2007) Comparative genomic analysis for the presence of potential enterococcal virulence factors in the probiotic Enterococcus faecalis strain Symbioflor 1. Int J Med Microbiol 297:533–539. https://doi.org/10.1016/j.ijmm.2007.02.008
Dubin K, Pamer EG (2017) Enterococci and their interactions with the intestinal microbiome. Microbiol Spectr. https://doi.org/10.1128/microbiolspec.bad-0014-2016
Dupuy B, Mani N, Katayama S, Sonenshein AL (2004) Transcription activation of a UV-inducible Clostridium perfringens bacteriocin gene by a novel σ factor. Mol Microbiol 55:1196–1206. https://doi.org/10.1111/j.1365-2958.2004.04456.x
Engels W, Siu J, van Schalkwijk S, Wesselink W, Jacobs S, Bachmann H (2022) Metabolic conversions by lactic acid bacteria during plant protein fermentations. Foods 11:1005. https://doi.org/10.3390/foods11071005
Escamilla-Martínez EE, Cisneros YMÁ, Fernández FJ, Quirasco-Baruch M, Ponce-Alquicira E (2017) Identification of structural and immunity genes of a class IIb bacteriocin encoded in the enterocin A operon of Enterococcus faecium strain MXVK29. J Food Prot 80:1851–1856. https://doi.org/10.4315/0362-028X.JFP-17-039
Escobar-Zepeda A, Sanchez-Flores A, Quirasco Baruch M (2016) Metagenomic analysis of a Mexican ripened cheese reveals a unique complex microbiota. Food Microbiol 57:116–127. https://doi.org/10.1016/j.fm.2016.02.004
Estrada K (2023) kjestradag/PAP: Initial release. https://doi.org/10.5281/ZENODO.7958138.
Flórez AB, Mayo B (2015) The plasmid complement of the cheese isolate Lactococcus garvieae IPLA 31405 revealed adaptation to the dairy environment. PLoS ONE 10:e0126101. https://doi.org/10.1371/journal.pone.0126101
Freitas AR, Pereira AP, Novais C, Peixe L (2021) Multidrug-resistant high-risk Enterococcus faecium clones: Can we really define them? Int J Antimicrob Agents 57:106227. https://doi.org/10.1016/j.ijantimicag.2020.106227
Ghaly TM, Chow L, Asher AJ, Waldron LS, Gillings MR (2017) Evolution of class 1 integrons: mobilization and dispersal via food-borne bacteria. PLoS ONE 12:e0179169. https://doi.org/10.1371/journal.pone.0179169
Hu C-B, Malaphan W, Zendo T, Nakayama J, Sonomoto K (2010) Enterocin X, a novel two-peptide bacteriocin from Enterococcus faecium KU-B5, has an antibacterial spectrum entirely different from those of its component peptides. Appl Environ Microbiol 76:4542–4545. https://doi.org/10.1128/AEM.02264-09
Ikhimiukor OO, Souza SSR, Marcovici MM, Nye GJ, Gibson R, Andam CP (2023) Leaky barriers to gene sharing between locally co-existing coagulase-negative Staphylococcus species. Commun Biol. https://doi.org/10.1038/s42003-023-04877-0
Kafil HS, Mobarez AM (2015) Spread of enterococcal surface protein in antibiotic resistant Enterococcus faecium and Enterococcus faecalis isolates from urinary tract infections. TOMICROJ 9:14–17. https://doi.org/10.2174/1874285801509010014
Kohl KD, Dieppa-Colón E, Goyco-Blas J et al (2022) Gut microbial ecology of five species of sympatric desert rodents in relation to herbivorous and insectivorous feeding strategies. Integr Comp Biol 62:237–251. https://doi.org/10.1093/icb/icac045
Liang W, Xie Y, Xiong W, Tang Y, Li G, Jiang X, Lu Y (2017) Anti-restriction protein, KlcAHS, promotes dissemination of carbapenem resistance. Front Cell Infect Microbiol. https://doi.org/10.3389/fcimb.2017.00150
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
Maldonado Galdeano C, Cazorla SI, Lemme Dumit JM, Vélez E, Perdigón G (2019) Beneficial effects of probiotic consumption on the immune system. Ann Nutr Metab 74:115–124. https://doi.org/10.1159/000496426
Markusková B, Lichvariková A, Szemes T, Koreňová J, Kuchta T, Drahovská H (2018) Genome analysis of lactic acid bacterial strains selected as potential starters for traditional Slovakian bryndza cheese. FEMS Microbiol Lett. https://doi.org/10.1093/femsle/fny257
Martino GP, Espariz M, Gallina Nizo G, Esteban L, Blancato VS, Magni C (2018) Safety assessment and functional properties of four enterococci strains isolated from regional Argentinean cheese. Int J Food Microbiol 277:1–9. https://doi.org/10.1016/j.ijfoodmicro.2018.04.012
Mendoza C (2019) Evaluación de marcadores genéticos para la diferenciación de cepas de Enterococcus de origen alimentario. Dissertation, School of Chemistry, Universidad Nacional Autónoma de México (UNAM), Mexico City.
Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M (2007) KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35:W182–W185. https://doi.org/10.1093/nar/gkm321
Néron B, Littner E, Haudiquet M, Perrin A, Cury J, Rocha E (2022) IntegronFinder 2.0: Identification and analysis of integrons across bacteria, with a focus on antibiotic resistance in Klebsiella. Microorganisms 10:700. https://doi.org/10.3390/microorganisms10040700
Nes IF, Brede DA, Diep DB (2013) Class II Non-lantibiotic bacteriocins. Handbook of Biologically Active Peptides (pp. 85–92). Elsevier. https://doi.org/10.1016/b978-0-12-385095-9.00016-6.
Nilsen T, Nes IF, Holo H (2003) Enterolysin A, a cell wall-degrading bacteriocin from Enterococcus faecalis LMG 2333. Appl Environ Microbiol 69:2975–2984. https://doi.org/10.1128/AEM.69.5.2975-2984.2003
O’Keeffe T, Hill C, Ross RP (1999) Characterization and heterologous expression of the genes encoding enterocin a production, immunity, and regulation in Enterococcus faecium DPC1146. Appl Environ Microbiol 65:1506–1515. https://doi.org/10.1128/aem.65.4.1506-1515.1999
Oliveira FS, da Silva RR, de Carvalho AF, Nero LA (2022) Genomic analyses of Pediococcus pentosaceus ST65ACC, a bacteriocinogenic strain isolated from artisanal raw-milk cheese. Probiotics Antimicrob Proteins 15:630–645. https://doi.org/10.1007/s12602-021-09894-1
Olvera-García M, Sanchez-Flores A, Quirasco Baruch M (2018) Genomic and functional characterisation of two Enterococcus strains isolated from Cotija cheese and their potential role in ripening. Appl Microbiol Biotechnol 102:2251–2267. https://doi.org/10.1007/s00253-018-8765-3
Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, Fookes M, Falush D, Keane JA, Parkhill J (2015) Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31:3691–3693. https://doi.org/10.1093/bioinformatics/btv421
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW (2015) CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. https://doi.org/10.1101/gr.186072.114
Prjibelski A, Antipov D, Meleshko D, Lapidus A, Korobeynikov A (2020) Using SPAdes de novo assembler. Curr Protoc Bioinform. https://doi.org/10.1002/cpbi.102
Qiao X, Du R, Wang Y, Han Y, Zhou Z (2020) Purification, characterization and mode of action of enterocin, a novel bacteriocin produced by Enterococcus faecium TJUQ1. Int J Biol Macromol 144:151–159. https://doi.org/10.1016/j.ijbiomac.2019.12.090
Quinn GA, Banat AM, Abdelhameed AM, Banat IM (2020) Streptomyces from traditional medicine: sources of new innovations in antibiotic discovery. J Med Microbiol 69:1040–1048. https://doi.org/10.1099/jmm.0.001232
Rahman MdS, Aktar U, Jani MR, Shatabda S (2018) iPro70-FMWin: identifying Sigma70 promoters using multiple windowing and minimal features. Mol Genet Genomics 294:69–84. https://doi.org/10.1007/s00438-018-1487-5
Rath D, Amlinger L, Rath A, Lundgren M (2015) The CRISPR-Cas immune system: biology, mechanisms and applications. Biochimie 117:119–128. https://doi.org/10.1016/j.biochi.2015.03.025
Ritz M, Garenaux A, Berge M, Federighi M (2009) Determination of rpoA as the most suitable internal control to study stress response in C. jejuni by RT-qPCR and application to oxidative stress. J Microbiol Methods 76:196–200. https://doi.org/10.1016/j.mimet.2008.10.014
Sambrook J, Fritsch EF, Maniatis T (2012) Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Sarantinopoulos P, Kalantzopoulos G, Tsakalidou E (2002) Effect of Enterococcus faecium on microbiological, physicochemical and sensory characteristics of Greek Feta cheese. Int J Food Microbiol 76:93–105. https://doi.org/10.1016/S0168-1605(02)00021-1
Savijoki K, Ingmer H, Varmanen P (2006) Proteolytic systems of lactic acid bacteria. Appl Microbiol Biotechnol 71:394–406. https://doi.org/10.1007/s00253-006-0427-1
Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. https://doi.org/10.1093/bioinformatics/btu153
Seipke RF, Kaltenpoth M, Hutchings MI (2012) Streptomyces as symbionts: an emerging and widespread theme? FEMS Microbiol Rev 36:862–876. https://doi.org/10.1111/j.1574-6976.2011.00313.x
Sun J, Lu F, Luo Y, Bie L, Xu L, Wang Y (2023) OrthoVenn3: an integrated platform for exploring and visualizing orthologous data across genomes. Nucleic Acids Res 51:W397–W403. https://doi.org/10.1093/nar/gkad313
Tao S, Chen H, Li N, Fang Y, Xu Y, Liang W (2022) Association of CRISPR-Cas system with the antibiotic resistance and virulence genes in nosocomial isolates of Enterococcus. IDR 15:6939–6949. https://doi.org/10.2147/IDR.S388354
van Heel AJ, de Jong A, Song C, Viel JH, Kok J, Kuipers OP (2018) BAGEL4: a user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res 46:W278–W281. https://doi.org/10.1093/nar/gky383
Vélez MA, Wolf VI, Espariz M, Acciarri G, Magni C, Hynes E, Perotti MC (2023) Study of volatile compounds profiles in milk matrices using Enterococcus faecalis EstA and Rhizomucor miehei lipase. Food Res Int 169:112861. https://doi.org/10.1016/j.foodres.2023.112861
Williams ML, Ghanem M (2022) Evaluation of candidate reference genes stability for gene expression analysis by reverse transcription qPCR in Clostridium perfringens. Sci Rep. https://doi.org/10.1038/s41598-022-23804-7
Wójcik W, Łukasiewicz M, Puppel K (2020) Biogenic amines: formation, action and toxicity—a review. J Sci Food Agric 101:2634–2640. https://doi.org/10.1002/jsfa.10928
World Health Organization (2017) WHO publishes list of bacteria for which new antibiotics are urgently needed. https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed. Accessed 5 July 2023.
Wright GD (2010) Antibiotic resistance in the environment: A link to the clinic? Curr Opin Microbiol 13:589–594. https://doi.org/10.1016/j.mib.2010.08.005
Wu Y, Pang X, Wu Y, Liu X, Zhang X (2022) Enterocins: classification, synthesis, antibacterial mechanisms and food applications. Mol 27:2258. https://doi.org/10.3390/molecules27072258
Zaidi S-Z, Zaheer R, Barbieri R, Cook SR, Hannon SJ, Booker CW, Church D, Van Domselaar G, Zovoilis A, McAllister TA (2022) Genomic characterization of Enterococcus hirae from beef cattle feedlots and associated environmental continuum. Front Microbiol. https://doi.org/10.3389/fmicb.2022.859990
Zheng J, Ge Q, Yan Y, Zhang X, Huang L, Yin Y (2023) dbCAN3: automated carbohydrate-active enzyme and substrate annotation. Nucleic Acids Res 51:W115–W121. https://doi.org/10.1093/nar/gkad328
Acknowledgements
This work was supported by UNAM-PAPIIT IN220921 and IN214423 grants. We thank Cindy A. Estrada Hernández for her support in RT-qPCR experimental procedures. María Elena Sánchez-Salazar edited the English manuscript.
Funding
This work was supported by UNAM-PAPIIT IN220921 and IN214423 grants.
Author information
Authors and Affiliations
Contributions
MQ contributed to the study’s conception and design. DIRS performed DNA extraction and sequence annotation analyses. DAP conducted the comparative genomics and sequence annotation analyses. SNFC conducted RNA extraction and transcription analyses. DAP and MQ produced the first manuscript draft. All authors commented on previous versions of the manuscript and read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Acero-Pimentel, D., Romero-Sánchez, D.I., Fuentes-Curiel, S.N. et al. Study of an Enterococcus faecium strain isolated from an artisanal Mexican cheese, whole-genome sequencing, comparative genomics, and bacteriocin expression. Antonie van Leeuwenhoek 117, 40 (2024). https://doi.org/10.1007/s10482-024-01938-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10482-024-01938-0