
Abstract
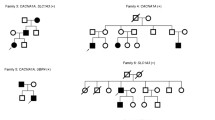
Mutations in the KCNA1 gene are known to cause episodic ataxia/myokymia syndrome type 1 (EA1). Here, we describe two families with unique presentations who were enrolled in an IRB-approved study, extensively phenotyped, and whole exome sequencing (WES) performed. Family 1 had a diagnosis of isolated cataplexy triggered by sudden physical exertion in multiple affected individuals with heterogeneous neurological findings. All enrolled affected members carried a KCNA1 c.941T>C (p.I314T) mutation. Family 2 had an 8-year-old patient with muscle spasms with rigidity for whom WES revealed a previously reported heterozygous missense mutation in KCNA1 c.677C>G (p.T226R), confirming the diagnosis of EA1 without ataxia. WES identified variants in KCNA1 that explain both phenotypes expanding the phenotypic spectrum of diseases associated with mutations of this gene. KCNA1 mutations should be considered in patients of all ages with episodic neurological phenotypes, even when ataxia is not present. This is an example of the power of genomic approaches to identify pathogenic mutations in unsuspected genes responsible for heterogeneous diseases.


Similar content being viewed by others

References
Baloh RW (2012) Episodic ataxias 1 and 2. Handb Clin Neurol 103:595–602
Graves TD, Cha YH, Hahn AF et al (2014) Episodic ataxia type 1: clinical characterization, quality of life and genotype-phenotype correlation. Brain J Neurol 137(Pt 4):1009–1018
Graves TD, Rajakulendran S, Zuberi SM et al (2010) Nongenetic factors influence severity of episodic ataxia type 1 in monozygotic twins. Neurology 75(4):367–372
Tan SV, Wraige E, Lascelles K, Bostock H (2013) Episodic ataxia type 1 without episodic ataxia: the diagnostic utility of nerve excitability studies in individuals with KCNA1 mutations. Dev Med Child Neurol 55(10):959–962
Browne DL, Gancher ST, Nutt JG et al (1994) Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet 8(2):136–140
Drmanac R, Sparks AB, Callow MJ et al (2010) Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327(5961):78–81
Zuberi SM, Eunson LH, Spauschus A et al (1999) A novel mutation in the human voltage-gated potassium channel gene (Kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain J Neurol 122(Pt 5):817–825
Kinali M, Jungbluth H, Eunson LH et al (2004) Expanding the phenotype of potassium channelopathy: severe neuromyotonia and skeletal deformities without prominent Episodic Ataxia. Neuromuscul Disord NMD 14(10):689–693
Kotagal V (2012) Acetazolamide-responsive ataxia. Semin Neurol 32(5):533–537
Lassche S, Lainez S, Bloem BR et al (2014) A novel KCNA1 mutation causing episodic ataxia type I. Muscle Nerve 50(2):289–291
Jan LY, Jan YN (1997) Cloned potassium channels from eukaryotes and prokaryotes. Annu Rev Neurosci 20:91–123
Zhu J, Alsaber R, Zhao J, Ribeiro-Hurley E, Thornhill WB (2012) Characterization of the Kv1.1 I262T and S342I mutations associated with episodic ataxia 1 with distinct phenotypes. Arch Biochem Biophys 524(2):99–105
van der Wijst J, Glaudemans B, Venselaar H et al (2010) Functional analysis of the Kv1.1 N255D mutation associated with autosomal dominant hypomagnesemia. J bBiol Chem 285(1):171–178
Long SB, Tao X, Campbell EB, MacKinnon R (2007) Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450(7168):376–382
Heeroma JH, Henneberger C, Rajakulendran S, Hanna MG, Schorge S, Kullmann DM (2009) Episodic ataxia type 1 mutations differentially affect neuronal excitability and transmitter release. Dis Model Mech 2(11–12):612–619
Haddad GA, Blunck R (2011) Mode shift of the voltage sensors in Shaker K+ channels is caused by energetic coupling to the pore domain. J Gen Physiol 137(5):455–472
Acknowledgments
The authors would like to thank the two families who volunteered for the research study. This work was supported by K08 AR055072 (PBA) from the National Institute of Arthritis and Musculoskeletal and Skeletal Diseases (NIAMS) of National Institute of Health (NIH), U19HD077671 (PBA and AHB) and P30 HD018655 from Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and National Human Genome Research Institute (NHGRI) of the NIH, the Research Connection at Boston Children’s Hospital (BCH), and The Manton Center for Orphan Disease Research at BCH.
Conflict of interest
The funding source had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. The authors have no conflict of interest to declare.
Author contributions
Drs. Agrawal and Brownstein had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Brownstein, Beggs, and Agrawal.
Acquisition of Data: Beggs, Rodan, Towne, Pelletier, Rosenberg, Urion, Picker, Cao, Tan, and Agrawal.
Analysis and interpretation of data: Brownstein and Agrawal.
Drafting of the manuscript: Brownstein, Pelletier, Beggs, Rosenberg, Towne, and Agrawal.
Critical revision of the manuscript for important intellectual content: all authors.
Statistical analysis: none needed.
Obtained funding: Agrawal and Beggs.
Administrative, technical, or material support: Beggs and Agrawal.
Study supervision: Beggs and Agrawal.
Funding/support
This work was supported by K08 AR055072 (PBA) from the National Institute of Arthritis and Musculoskeletal and Skeletal Diseases (NIAMS) of National Institute of Health (NIH), U19HD077671 (PBA and AHB), and P30 HD018655 from Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and National Human Genome Research Institute (NHGRI) of the NIH, the Research Connection at Boston Children’s Hospital (BCH), GETTYLAB, and The Manton Center for Orphan Disease Research at BCH.
Role of funder/sponsor
The funding source had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Brownstein, C.A., Beggs, A.H., Rodan, L. et al. Clinical heterogeneity associated with KCNA1 mutations include cataplexy and nonataxic presentations. Neurogenetics 17, 11–16 (2016). https://doi.org/10.1007/s10048-015-0460-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-015-0460-2