
Abstract
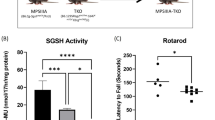
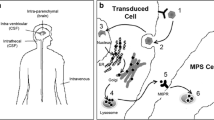
The mucopolysaccharidoses are a group of lysosomal storage diseases caused by deficiency of an enzyme required for the normal degradation of glycosaminoglycans. Patients with mucopolysaccharidosis typically have widespread lysosomal storage, skeletal and central nervous system disease, and hepatosplenomegaly. Some patients with mucopolysaccharidosis may benefit from enzyme replacement therapy or bone marrow transplantation. Animal models of mucopolysaccharidosis have proven valuable for the evaluation of the effectiveness of potential treatments for patients with lysosomal storage disease. A murine model of MPS VII (Sly syndrome) has proven particularly useful because of its well-defined genetics and its well-characterized clinical, pathologic, and biochemical alterations, which resemble those seen in patients with mucopolysaccharidosis. Correction of these alterations forms the basis for evaluation of the effectiveness of novel treatments. A wide range of therapies have been tested using this model, including enzyme replacement therapy, bone marrow, stem cell, and neural progenitor cell transplantation, and a variety of viral-mediated gene therapies. The inferences drawn from these therapeutic studies using the murine MPS VII model are likely generalizable to other lysosomal storage diseases.
Similar content being viewed by others

Author information
Authors and Affiliations
Additional information
Received March 10, 2001; accepted April 5, 2001.
Rights and permissions
About this article
Cite this article
Vogler, C., Barker, J., Sands, M. et al. Murine Mucopolysaccharidosis VII: Impact of Therapies on the Phenotype, Clinical Course, and Pathology in a Model of a Lysosomal Storage Disease. Pediatr. Dev. Pathol. 4, 421–433 (2001). https://doi.org/10.1007/s10024001-0079-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10024001-0079-1