
Abstract
Metal surfaces covered with oxides have attracted considerable scientific attention in various applications. In particular, anodic films fabricated by cost-effective anodizing have been widely used in nano-structured engineering to provide various surface functionalities. However, understanding of alloy film stability, having individual elements with widely varying structures and morphologies, is very limited due to lack of thermodynamic information and effects of electrolyte chemistry. This requires many tedious efforts on a trial and error basis in selecting suitable electrolytes that can produce the protective film at high efficiency on alloys having mixed chemistries. It is, therefore, crucial to develop a combination of high throughput theoretical analysis and automated rapid localized electrochemical probing that provides a fast and simple solution for electrolyte choice and paves the way to the remarkable expansion of industrial applications of oxides. Herein, we demonstrate that combinatorial Al–Gd alloys covering 1.0 to 10.0 at.% Gd can be oxidized into ultra-thin anodic films of controlled thickness through a selection of electrolyte based on thermodynamics (phosphate buffer with a pH of 8.20). We propose that growth of anodic films on alloys at high efficiency is possible if Gibbs free energy minimization criteria would be systematically contemplate.
Graphical abstract

Similar content being viewed by others

Introduction
The “1-to-1” approach involving one system, one calculation, and one experiment at a time is now regarded as the traditional, time consuming, and outdated method in materials research, when there is an ever-increasing need for strategies to develop advanced materials with novel functionalities [1, 2]. The intelligent strategies play a crucial role in technologies ranging from electronics, energy generation and storage, aerospace, medicine, and defence industry [3,4,5]. To achieve these technologies, attention has been given recently to integrate high-throughput first-principles calculations and combinatorial methods to obtain robust, extrapolative tools for gaining insights into structure–property–composition relationships in order to reduce the experimental efforts, time, and cost in the materials discovery process [6,7,8,9,10,11,12,13].
Ab initio or first-principles computational methods are becoming important in new materials discovery [14]. Scheming fabrication strategies using high-throughput computational approaches provide numerous benefits over traditional experimental-based materials discovery methods. Estimating material’s fundamental properties is often faster with computation tools, when compared to the experiment, and properties of thousands of materials can be calculated in a relatively short period of time. Additionally, it is often trivial in calculations to simulate the effects of chemical substitutions or lattice strain, but achieving these through experiments could take many months of painstaking laboratory work. Thus, computations are effective tools for designing new materials [15,16,17]. Directly linked to this, combinatorial approaches are defined by the development of a “library” that comprises a material parameter of interest (usually dependent on the gradient in composition) addressed by rapid, local, and automated measurements creating a large data set. These data sets are examined to find a trend between composition, structure, and property of interest. In other words, combinatorial methods are generally faster and less expensive than traditional “1-to-1” material fabrication approaches. Furthermore, since all experiments are performed on the same graded-library with the same measurement tools and identical specimen preparation and storage history, a comprehensive and reliable data set can be generated [1, 18, 19].
The applications of these intelligent strategies are important, in particular, to rapidly explore the lesser-known and rarely explored rare-earth elements (REEs) and their alloys. The understanding and fabrication of REEs and their alloys through the electrochemical route are very limited due to the lack of both experimental and theoretical literature. As an example, very few efforts have been made to explore the properties of the central atom of lanthanoid series, the gadolinium (Gd), and its oxide (Gd2O3). Gadolinium ortho-aluminate (GdAlO3) mixed oxide possesses excellent magnetic and antiferromagnetic properties [20,21,22]. The pure oxide Gd2O3 is a wide bandgap material (5.37–6.37 eV) [23] and meets the low leakage current requirement in CMOS applications [24]. The superior optical properties of Gd2O3 thin films make them efficient for use in optical applications [23, 25, 26]. Hong et al. reported that Gd2O3 is an excellent dielectric material to passivate GaAs surface, with a dielectric constant of 10 for epitaxial cubic phase and a low leakage current density of 10−9–10−10 A cm−2 at zero bias [27], while Zhou et al. have found out a value of 20 for Gd2O3 thin-film [28]. This indicates that changing the substrate and growth method affects the anisotropic polycrystalline film growth, which can enhance its dielectric constant. The dielectric constant of Gd2O3 can be further used to modify the dielectric behaviour of aluminium-based alloys. The dielectric constant is one of the factors responsible for capacitance enhancement, and portable electronic devices have increased the demand for small-size electrolytic capacitors with high capacitance, low equivalent series resistance, and low leakage current [29, 30]. Materials with high dielectric constants are also crucial in many areas of modern microelectronics for miniature supercapacitors, infrared detectors, radar absorbing coatings, and holographic devices. Thus, there is a growing interest in the synthesis and precise characterization of materials with high dielectric constants and low leakage currents.
The comprehensive understanding of oxide behaviour in a solution can be enhanced by combining theoretical predictions with localized measurements. The localized analysis is critical as the dielectric properties are generally dependent on the material geometries and crystalline orientations [31]. At microscale, properties may be very different than those at macroscale. It is therefore crucial to probe these properties at a scale where inhomogeneities may be specially resolved. The development of scanning droplet cell microscopy (SDCM), in particular, has allowed for spatially resolved electrochemical characterization of surfaces [32,33,34]. Coupling SDCM with electrochemical impedance spectroscopy (EIS) may even determine the grain-specific responses and has superior control of the working area [31,32,33,34,35,36].
In this study, we take advantage of computational and combinatorial strategies to explore the composition, structure, and properties relationship systematically in Al-Gd alloys. For this purpose, the predictions of possible stable phases in a given electrochemical system were obtained from conventional potential-pH (or E-pH) diagrams and DFT simulated compositional phase diagrams and E-pH diagrams. This permits a rapid identification of anodizing electrolytes over a wide pH range for fabrication of Al–Gd alloys in an ultra-fast manner and calculations of material specific parameters.
Experimental methods
Laterally graded Al–Gd alloy library was obtained by thermal co-deposition of the Al and Gd from two separated sources on borosilicate float glass substrates (VWR International GmbH, Germany) having dimensions of 2.6 × 7.6 cm2. A thermal co-evaporator with a base pressure of 10−5 Pa was used for co-deposition of Al and Gd. The compositional control along the library was obtained by individually adjusting the deposition rate of each source. Specific details of the hardware used for library deposition were previously reported [37]. A low Gd amount was desired along the library, thus deposition rates of 0.9 and 0.1 nm s−1 were used for Al and Gd, respectively, to obtain a compositional spread of approximately 9 at.% along the length of the substrate. A total film thickness of 350 nm was obtained at the middle of Al–Gd library. It is worth mentioning here that the compositional spread of a library is limited by the particularities of the cosine law governing the thickness uniformity of alloying element taken separately along the substrate. For Gd contents above 10 at.%, a new compositional spread needs to be prepared. However, higher Gd contents would result in formation of various intermetallics, which is not desired here.
Immediately after Al–Gd library deposition, scanning energy-dispersive X-ray (SEDX) spectroscopy was used for compositional mapping of the Al–Gd spread. Each alloy was measured at discreet surface locations and their quantitative analysis was performed using IDFix software (remX GmbH, France). The Gd amount varied between 1.0 and 10.0 at.% along the library, while the Al concentration varied complementarily between 99.0 and 90.0 at.%. This defines a compositional resolution of the Al–Gd thin film library of 0.12 at.% mm−1. For an usual requirement of 1 at.% precision for alloy identification, more than 7 mm are thus available on the surface of the library for characterizing a single Al–Gd alloy. This resulting area is quite convenient for being addressed by other investigation techniques for Al–Gd alloy property mappings. X-ray diffraction (XRD) was used to map the crystallographic particularities of Al–Gd alloys as a function of their composition. The experiments were performed in θ–2θ (Bragg–Brentano) and ω-2θ with ω = 3o (grazing incidence) geometries with CuKα radiation. Additionally, the surface morphologies of individual Al–Gd alloys (defined by EDX mapping) were characterized by scanning electron microscope (SEM—Zeiss Gemini 1540 XB) before and after anodization.
The electrochemical measurements on thermally co-deposited Al–Gd films were carried out using the SDCM in contact mode [32] at ambient temperature and pressure. The SDCM is a three-electrode system containing two channels in V-geometry, serving as inlet and outlet for the electrolyte. A Pt wire positioned in the electrolyte supply compartment served as counter electrode while a Hg/Hg2Cl2 µ-reference electrode was placed in the drain compartment for avoiding chloride contaminations [38]. The Al–Gd thin film library deposited on glass substrates served as working electrode. The addressed area (0.049 cm2) was determined by SEM examination of anodically grown films. A steady flow of electrolyte is provided by a peristaltic pump that works as both electrolyte supply and drain at a constant rate.
All measurements were carried out using self-developed Labview software that controls the scanning of the SDCM tip along the library (using an XY translation stage) and monitors the applied force during contact. In this way, a single droplet of electrolyte is moved along the surface. At each unique XY position which also corresponds to unique alloy composition, open-circuit potential (OCP) was measured for 180 s in 0.2 M phosphate buffer having a pH value of 8.20 (Fig. 1). The buffer solution was obtained by mixing anhydrous potassium phosphate monobasic and sodium phosphate dibasic heptahydrate. Following OCP recording, Al–Gd alloys were locally anodized potentiodynamically to 10 V. Film resistance and dielectric properties of grown films were determined by simultaneously coupling EIS with cyclic voltammograms (CV) using a Compact Stat electrochemical interface system (IVIUM Technology, The Netherlands) with an AC amplitude of 50 mV in a frequency range of 105 to 10−1 Hz. All impedance spectra in the present study were recorded at 0 V vs. SHE. The present study was conducted using the constant electrolyte flow from inlet to outlet compartments within the SDCM body. This ensured fresh electrolyte at the contact with the working electrode during combined CV-EIS measurements. The protocols followed in the electrochemical study is as follows: (1) positioning of cell at a given location on the substrate and OCP recording, (2) anodic sweep (100 mV s−1) from OCP value measured before, (3) EIS measurement of grown film, (4) looping of (1—3) to 10 V at 1 V incremental step. A more detailed description of the microelectrochemical flow measurement can be found elsewhere [6, 32, 39]. The thermodynamic data and electrochemical reactions used in the present study were taken from different sources [40,41,42,43,44,45,46]. Information regarding formation energies of species was taken from an open source web application [47]. The E-pH diagrams from first-principles DFT calculations were constructed from the high-throughput infrastructure developed by Jain et al. [46, 47], where experimentally determined free energies of aqueous ions and DFT energies for solid phases were used to construct multi-element compositional phase diagrams and E-pH diagrams. First-principles Car-Parinello molecular dynamics was used for simulations of aqueous states [48]. All E-pH diagrams were calculated at 298 K and atmospheric pressure for dissolved concentrations of species per litre of water.

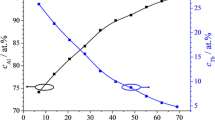
Open circuit potential (OCP) of compositionally graded Al–Gd alloy library in phosphate buffer having pH 8.20 for 180 s
Results and discussion
Film structure and morphology
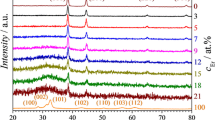
The crystallographic properties of individual alloys were examined in both θ–2θ and ω-2θ modes along the Al–Gd library. XRD analysis was performed at various Al:Gd ratios and selected XRD patterns are presented in Fig. 2 as a function of Gd concentration. Aluminium possesses fcc symmetry, whereas Gd has a hexagonal structure. Additional phases other than Al were not found and the addition of Gd did not induce tri-aluminide intermetallic phase. Significant reduction in Al(111) peak intensity is observed when mapping from higher to lower Gd concentration. This could be due to variation in Al and Gd ratios as well as the gradual loss of crystallinity which indicates the influence of Gd concentration.

XRD pattern of thermally evaporated Al–Gd combinatorial alloys ranging from 1.5 to 10.0 at.% Gd
The binary Al–RE phase diagram and detailed analysis of Al–RE systems by Gschneidner et al. [49] indicate that GdAl3 tri-aluminide intermetallic phase is formed between 0 and 25 at.% Gd addition to the Al matrix. The formation of the REAl3 phase is linked with the transformation of the structure from predominately hexagonal to cubic for the light lanthanoids having large radii (La, Ce, Nd, Pr, Sm, and Gd). Contrary to binary Al–Gd phase diagram and Gschneidner et al. prediction, the absence of the GdAl3 phase may be attributed to the low sensitivity of the XRD analysis at nm-depth scale. Additionally, Al rich-phase predominates in the alloy matrix, and it is likely that the presence of GdAl3 is eclipsed by the Al-rich matrix.
The surface morphologies along the Al–Gd compositional spreads before and after electrochemical studies were examined by SEM. The SEM images prior to anodizing were taken in the vicinity of anodizing spot for each composition. Figure 3 shows the map of the observed morphologies from 1.5 to 10 at.% Gd, and the composition of each investigated region is given at the upper left corner. The microstructural analysis discloses a gradual change in surface morphology as the Gd content in the Al matrix increases (Fig. 3a) and can be divided into three distinct parts. The typical Al grains can be seen in the first line of SEM images, ranging from 1.5 to 2.5 at.% Gd. The suppression of large grains and gradual formation of mixed morphology are evident from 3.5 to 5.5 at.% Gd (second line), where the imprint of surface grains and grain boundaries still can be seen. A noticeable change in the surface morphology and distinct features with thick upright needles can be observed from 7.0 to 10 at.% Gd. Moreover, the change in surface features with Gd addition agrees well with XRD analysis, where the reduction in peak intensity and loss of crystallinity were attributed to variation of the Al:Gd ratio.

Surface morphologies along Al–Gd alloy library a before and b after anodizing at selected atomic concentrations of Gd. The Gd concentration in the alloy is given at the upper left corner of each figure. The SEM images were taken after room temperature anodizing at a sweep rate of 100 mV s−1 to 10 V in 0.2 M phosphate buffer having a pH value of 8.20
Surface morphologies after anodizing reveal a significant change and show a network of bright lines on the original Al grains, as can be readily seen from the SEM image in Fig. 3b. Because of their strong adsorption on the metal surface, the possibility of phosphate precipitates cannot be overlooked. No signs of localized corrosion or pitting was found in SEM observation.
Electrochemical studies
Composition and structural complexity in binary Al–RE alloys render the electrochemical measurement unstable [50]. One alloy constituent may dissolve preferentially in a certain pH range leaving the other constituent unaffected. Structure and morphological complexity due to lattice mismatch and a large difference in atomic and ionic radii further complicate the understanding of dissolution susceptibility in a specific pH range. Consequently, predicting thermodynamically auspicious conditions for anodizing multi-component systems is a meticulous task due to a large number of competing reaction pathways and end products. Additionally, the classical E-pH diagrams do not address the alloy–H2O systems, thereby producing a gap in the theoretical understanding of Al–RE alloys [40, 41, 51, 52]. Hence, a theoretical framework is also developed in the present study, which can readily foresee the thermodynamic feasibility of reactions, stable phases in a complex Al–Gd–P–O–H system. For instance, conventional E-pH diagrams do not reveal the presence of AlPO4, and GdPO4 phases in Al–H2O and Gd–H2O aqueous systems. Consequently, calculations of material specific parameters and analysis of film properties would remain incomplete without considering missing phases in conventional E-pH diagrams.
In order to find a compromise pH range for Al–Gd alloys in phosphate buffer, the conventional E-pH diagrams Al–H2O, Gd–H2O, P–H2O systems were simulated from an experimental database [40,41,42,43,44] and superimposed on each other. Figure 4a shows the superimposition of Al–H2O and Gd–H2O systems, while Fig. 4b reveals the superimposition of Gd–H2O and P–H2O systems. A different stability domain for Gd(OH)3 and Gd2O3 arises because of the difference in free energies for hydroxide and oxide formations (see Table 1). In other words, considering the metal hydroxide alone is not sufficient to predict the pH range even for a single metal. Figure 4a discloses that the domain of Al2O3 stability terminates at around pH 9.0 and shifts towards lower pH range as the dissolved Al concentration in the solution reduces. Additionally, passive domains in the Gd–H2O system begin at around pH 9.0 and moves towards higher pH values when the concentration of dissolved Gd is decreased in the solution. Simultaneous oxidation of Al and Gd alloys at high efficiency or at a nearly equal amount of dissolved concentrations of both metals is not thermodynamically feasible in the aqueous electrolyte at any pH value. Consequently, oxide-forming species such as phosphate and borate are needed to shift the passive domains of both metals. However, it was further found out that superimposing conventional pH diagrams of metal–H2O systems with oxide-forming species does not modify the E-pH diagrams. One reason for this is that superimposed diagrams do not incorporate the reactions between metals and oxide-forming species. Thus, it is not possible to progress in the understanding of an alloy anodizing in a complex aqueous medium by just superimposing a series of conventional Pourbaix diagrams, and this may often lead to false conclusions in predicting aqueous corrosion. Moreover, extracting thermodynamic information from published literature is a very time-consuming effort. Many alloys exhibit intriguing physical and chemical properties resulting from the complex interactions between the constituent’s elements. Several known or unknown non-equilibrium compositions and structures can form in a material during its lifetime and at present, it is challenging to accurately simulate all of the relevant phases in a given system. In order to overcome this limitation, there is a need for an advanced approach to predict the realistic complex systems in a high-throughput manner. Therefore, to maximize the prediction of possible phases in the Al–Gd–P–H–O complex system, the grand canonical linear programming (GCLP) method [7, 9] and open-source web application [45,46,47] were also utilized for analysing the complex ground state thermodynamics. GCLP strategy works by mapping a free energy minimization problem as a linear algebra problem and this allows the computation of very large number of phases in a very high-dimension phase space. The exemplary DFT simulated compositional phase diagrams for the identification of stable or metastable phases in Al–Gd–P–O2–H2 system are provided in the supplementary Figures S1, S2 and S3. An example of implementation of above-mentioned approach to construct E-pH diagrams can be seen in Fig. 5, where stable phases found in compositional phase diagrams of Al–Gd–P–O2–H2 system can be considered for E-pH diagrams of Al–P–H2O (Fig. 5a), Gd–P–H2O (Fig. 5b), Al–Gd–P–H2O (Fig. 5c) aqueous systems. Figure 5d reveals the summary of enthalpy change estimated from DFT calculations for Gd2O3–H2O, Al2O3–H2O, Al2O3–Gd2O3, Gd2O3–P2O5, and Al2O3–P2O5 binary systems. It is obvious that phosphate incorporation remarkably modifies the E-pH diagrams of both constituents of the alloy and provides the protection of Al3+ and Gd3+ species around pH 6.0–9.0 (Fig. 5c) above 0 V. Consequently, phosphate-containing electrolyte was chosen between pH 6.0 and 9.0. The oxide-forming species such as phosphate cannot be selected by just considering the free energies of species (AlPO4 and GdPO4). There must be some thermodynamic pH range at which a specific electrolyte can be used. It is worth-mentioning here that the main objective of superimposed E-pH diagram computations was to identify the electrolyte and pH ranges. A similar framework can be designed for other electrolyte selection such as phosphate-citrate buffer, ammonium pentaborate tetrahydrate.

a Superimposed E-pH diagram of Al–H2O and Gd–H2O systems. The diagrams are constructed at different molar concentrations of dissolved Al and Gd with exponents of 0, −3, −6, and −8 in water at 298 K; b superimposed E-pH diagram of Gd–H2O and P-H2O systems for Gd2O3/Gd(OH)3 formation at molar concentration per litre of water with exponent of −6
Following theoretical consideration and electrolyte selection, schematic and expected mixed oxide growth for the present study is shown in Fig. 6, which discloses the use of SDCM along Al–Gd library (a), oxidation of Al and Gd atoms upon application of formation voltage, V and interaction of cations with the phosphate buffer (b, c). The relative sizes of atomic and ionic radii of Al and Gd are noticeable before and after the application of V as the migration rate of cations through the film depends on their sizes and bond energies. The oxidation of alloying elements in Al-binary alloys depends on Gibbs free energy per equivalent for the formation of the alloying element oxide, relative to that of formation of alumina. A more negative free energy value of the alloying element oxide relative to alumina specifies that the alloying element has a higher mobility than Al [53,54,55,56,57]. As Gd2O3 and GdPO4 have higher negative free energy compared with Al2O3 and AlPO4, it is likely that Gd3+ will migrate faster than Al3+ during anodizing Al–Gd alloys, and both Al and Gd ions would be incorporated into the oxide film and would form units of oxides. Phosphate adsorption on the surface of mixed oxide is suggested in part (d) of the figure, where the expected amorphous character of the anodic film is neglected in order to better suggest mixing. The mixing from the molecular point of view is represented in part (e). Very large sizes of AlPO4 and GdPO4 molecules and closed structure of mixed oxide (GdAlO3) suggest that both AlPO4 and GdPO4 despite their thermodynamic stability may have limited inward migration rate. Because of the constant flow of fresh electrolyte, it is assumed that phosphate-containing species will flush out leaving only a minute quantity of \({\mathrm{PO}}_{4}^{3-}\) on the film surface as adsorbed species.

Simulated E-pH diagrams using first-principles DFT methods: a P–Al–H2O system at \({a}_{{\mathrm{Al}}^{3+}}= {10}^{-6}\mathrm{M},\) \({a}_{{\mathrm{PO}}_{4}^{3-}}= {10}^{-1 }\mathrm{M}\); b P–Gd–H2O system at \({a}_{{\mathrm{Gd}}^{3+}}={ 10}^{-6}\mathrm{M},\) \({a}_{{\mathrm{PO}}_{4}^{3-}}= {10}^{-1 }\mathrm{M}\); c P–Al–Gd–H2O system at \({{a}_{{\mathrm{Al}}^{3+}}= a}_{{\mathrm{Gd}}^{3+}}= { 10}^{-6}\mathrm{M},\) \({a}_{{\mathrm{PO}}_{4}^{3-}}= {10}^{-1 }\mathrm{M}\). d Enthalpy change of oxide formation for Gd2O3–H2O, Gd2O3–P2O5, Gd2O3–Al2O3, H2O–Al2O3, and P2O5–Al2O3 binary oxide phase diagrams using first-principles DFT methods
Potentiodynamic oxide formation
Anodizing and EIS studies were performed on alloys along the library. Series of cyclic voltammograms with a step-wise increase of V between 1 and 10 V were recorded at 11 different compositions by positioning the tip of SDCM along the Al–Gd compositional spread. The graphical representation of selected CVs is presented in Fig. 7 which corresponds to 110 potentiodynamic scans and EIS spectra. For each composition, a visible current density plateau can be readily observed which defines the charge consumed during the incremental growth process. This is due to the direct relationship between the charge transferred to the working electrode and the oxide volume grown following Faraday’s law. This leads to the average oxide formation factor, kave which can be defined as
where \({K}_{\mathrm{ox}}\) is a material constant, d is the oxide thickness, V is anodizing potential, q is the charge consumed, and η is the efficiency of the film formation current. \({K}_{\mathrm{ox}}\) can be defined in terms of oxide specific constants (mixed oxide molar mass, \({M}_{\mathrm{ox}}\), oxide density,\({\rho }_{\mathrm{ox}}\), and oxidation number, z, and Faraday’s constant F) by the following equation:

Schematic of mixed oxide formation: a use of SDCM on evaporated film surface; b, c interaction of mixed-structure with phosphate-containing aqueous electrolyte; d formation of mixed oxide and phosphate adsorption on film surface; e molecular structure of mixed oxide (GdAlO3) and metal phosphates (AlPO4 and GdPO4)
The molecular masses and densities of mixed anodic films locally grown on the Al–Gd alloys library were estimated using the mixed-matter theory by means of linear distributions between the Al2O3 and Gd2O3. Owing to negligible dissolution of Al and RE in phosphate-containing electrolytes [58,59,60,61,62,63,64,65], a typical passive film current density plateau and negligible leakage current density response on reversal of potential sweep scans, the efficiency of film formation current η can be assumed here as 100%. Because of the large sizes of GdPO4 and AlPO4, it is expected that phosphate incorporation within the film would be extremely small. The extremely low dissolution of RE in phosphate-containing electrolyte was verified by coupling CV-EIS measurements and ICP-OES analysis in Al–Ho system (~ 10–20 ppb) [64]. The current overshoots present at low potentials may be attributed to the delayed oxide growth due to the space charge layer formation or the sudden reaction of the metal surface to the electrolyte. Therefore, the CV and EIS data are excluded at 1 V in kave calculation.
Electrochemical impedance spectroscopy
Bode and Nyquist plots for two extreme compositions are shown in Fig. 8. The impedance changes linearly as a function of frequency for all compositions, and gradually rises with V implying a linear growth of the anodic film. The direction of arrows shows the increase in anodizing potential. As the film grows, an expected shift of phase angle towards − 90o typical of dielectric oxide film is obtained. The value of phase shift, α obtained from impedance fitting was above 0.92 in all compositions.

CVs at different atomic concentrations of Gd along the Al–Gd library. Each single alloy composition was electrochemically scanned in phosphate buffer (pH 8.20) from 0 V to a maximum of 10 V and then scanned back to 0 V. The V was stepwise increased with 1 V increment
Figures 8a, c show that regardless of film composition, Bode plots on a log scale show similar absolute impedance. The corresponding Nyquist plots on a linear scale in Fig. 8b, d disclose the magnitude reduction in the imaginary component (-Zimg) of impedance without affecting the real component (Zr). This indiactes that Gd addition affects the capacitance or dielectric properties and has no appreciable effect on the resistance of passive films. As the capacitance varies inversely with the impedance, it is envisaged that films comprising Al and Gd mixed oxides can be obtained by incorporating Gd with enhanced capacitance into Al matrix while retaining the film resistance, Rf of pure alumina. Moreover, the phase shift behaviourr reveals that as oxide grows thicker, incremental V not only induces a capacitive response, as evidenced by the magnitude of phase shift, but also widens the constant phase element (CPE).
The influence of Gd on dielctric constant can be obtained by a careful determination of capacitance through EIS fitting. Different approaches are used in the literature to calculate the true value of capacitance from CPE without considering the origin of the CPE [66,67,68]. It is difficult to implement a suitable CPE-C conversion model, thereby, the determination of capacitance from CPE data is often inadequate, leading to the erroneous prediction of physical properties [69,70,71]. For this reason, special consideration has been taken to understand the CPE behaviour for passive film growth.
The impedance of Rel(QRf) circuit exhibiting CPE behaviour having parameters Q and α can be expressed as
where Rel and Rf are the electrolyte and film resistances, respectively. Hirschorn et al. demonstrated the significance of employing the correct formula for a given distribution of time constants as well as impact on film properties [69]. Hsu and Mansfeld [67], on the other hand, considered the frequency, ωmax on the top of the depressed semi-circle where the real part (Zr) lies midway between low and high-frequency x-axis intercepts on the Nyquist plot. As a result, multiple approaches are compared in order to accurately predict the capacitance, C and kave from CPE having parameters Q and α. The results for the kave-values were also compared with the kave values obtained from Faraday’s law (Table 2). The Rf, α, and Q values used in Table 2 were obtained by analysing the EIS data using ZView software.
It is also worth mentioning here that Hsu-Mansfeld and Hirschhorn et al. presumed the semi-circle depression model while deriving the mathematical expression for CPE without considering the role of film resistance, Rf in Eq. (3) [67, 69]. Obviously, the term “Q(jω)α” in Eq. (3) cannot completely determine the resultant impedance of Rel(QRf) circuit, and CPE behaviour in itself cannot be fully understood by neglecting Rf. Therefore, to understand the uniform film growth as a function of V, the impedance spectra are also simulated in parallel by assuming different scenarios as illustrated in Fig. 9.

Bode and Nyquist plots recorded during simultaneous CV and EIS measurements along the Al–Gd alloy library during film growth for a, b Al–2.0 at. % Gd alloy, c, d Al–9.5 at.% Gd alloy. The data were recorded after each 1 V incremental in CV to a maximum of 10 V

Simulated Nyquist and Bode plots: a α varies, b α and Rf varies, c α, Rf, Q changes. The electrolyte resistance Rel is kept constant for these simulations

a Inverse capacitance, C−1 as a function of V b relative permittivity, \({\varepsilon }_{r}\) from Faraday’s law. The EIS data is estimated using ZView software
Case 1 in Fig. 9a illustrates the situation where only α changes uniformly from 1.0 to 0.5 as commonly observed in the semi-circle depression model. Changing α without affecting Q and Rf does not replicate the typical impedance spectra as generated in uniform film growth (see Fig. 8) and even suppresses the CPE behaviour when α approaches 1.0 as demonstrated in the phase shift vs. frequency plot. Case 2 describes the resistive film scenario in which the film resistance, Rf and the ratio Rf/Rel vary considerably, and CPE parameters Q and α contribution are largely suppressed due to the dominating effect of Rf (Fig. 9b). Under such conditions, the CPE behaviour starts extending towards the lower frequency part of impedance spectra. Finally, case 3 represents the most realistic situation of thin-film growth by anodizing where all α, Rf, and Q vary in a cooperative manner during film growth (Fig. 9c). The widening of the CPE towards low or high -frequency end of Bode plot will largely depend on the dominant effect from Q and Rf. The CPE broadening for the highly capacitive film, must occur towards the higher frequency side, while for a resistive film having negligible contribution of Q, it should be extended towards low frequency. Lastly, uniform film growth can be easily understood from the shape of the Bode plot where Rf, Q, and α are changing as a function of V in the present study.
The summary of EIS analysis is provided in Fig. 10 and Table 2 in the form of inverse capacitance, C−1, kave, and relative permittivity, \({\varepsilon }_{r}\) as a function of V. The C−1 increases linearly with V (Fig. 10a), as expected from the following equation:
in which, \({\varepsilon }_{o}\) is the permittivity of vacuum and A is the surface area.
Comparison of oxide formation factors
Table 2 indicates that EIS analysis yields kave values ranging from 1.34 to 1.62, while the kave values vary from 1.52 to 1.76 when calculated from Faraday’s law. Comparison of k-factor obtained from different equations reveals that Hsu-Mansfeld equation (Cox,M), Faraday’s law, and normal distribution approach (Cox,n) yield similar values of true capacitance. Additionally, the surface distribution (Cox,s) akin to Brug’s formula [68] gives an unrealistically high estimation of kave values. As the contribution of Rf is negligible in the surface distribution approach, it is probable that the approach will give an erroneous prediction of kave value when applied to the passive oxide film. Slightly higher kave values are obtained from the Faraday’s law as the kave from EIS measurement involves the calculation of film thickness directly resulting from the film, while Faraday’s law requires the anodizing charge (including side reactions) to be used directly for the determination of d values. The additional charge consumed in side reactions such as oxygen gas evolution and metal dissolution affects the kave. Moreover, the mixed matter approach was assumed for the densities and molecular masses of the locally grown oxide film. As the few nm thin films are difficult to characterize, the errors in kave values from EIS and Faraday’s methods are inevitable. Precise assumptions of material constants, accurate Cox estimation, and negligible side reactions would ensure comparable kave values from EIS and Faraday’s law.
Dielectric constant
The εr (Fig. 10b) was determined from the slope of V plotted against C−1. Figure 10b shows that \({\varepsilon }_{r}\) values are ranging from 13.5 to 23.0 for Al–Gd alloys indicating that addition of Gd to Al improves the dielectric constant of pure alumina. Because of material’s constant assumption in mixed matter theory, a 10% error bar is also included in Fig. 10b. \({\varepsilon }_{r}\) of anodic films containing mixed oxides lies between the values reported for pure Al2O3 (εr for Al2O3 = 9–13) and pure Gd2O3 (\({\varepsilon }_{r}\) for Gd2O3 = 10–24 on single and polycrystalline thin-films) [24, 27]. \({\varepsilon }_{r}\) values of both Al2O3 and Gd2O3 in phosphate buffer (pH 8.20) using SDCM approach were found out to be 12.7 and 20.5 respectively. This result is a prominent example of the advantage of combinatorial investigations over common batch specimen production, where the property of interest (\({\varepsilon }_{r}\) in the present case) can be readily identified for wide range of compositions with high accuracy, since the history of preparation, surface treatment, air contact, and other steps are identical. The variation of the screened property can be solely attributed to the difference in composition.
The efficiency of film formation, composition, and nature of amorphous and crystalline phases within the mixed structure can be estimated by a combination of FIB-TEM, XPS, RBS analyses which in the present case is not possible owing to the ultra-thin anodic films. At such a low value (~ 1–15 nm), obtaining a smooth RBS spectrum and its further simulation is a very challenging task. No visible sign of gas evolution, extremely low dissolution of rare-earth elements in phosphate buffer solution, and detection of negligible Ho (14 ppb) using the SDCM-ICPSOES approach indicates the negligible consumption of ionic current in various side reactions [59, 64].
Conclusions
In summary, a combination of a high-throughput simulation and high-throughput scanning was investigated in a chosen electrolyte on an Al–Gd compositional spread covering a range from 1.0 to 10. at.% Gd. The results of the present study must be interpreted in terms of used electrolyte and special SDCM geometry. The main conclusions are as follows:
-
1.
Novel E-pH and phase diagrams were simulated by coupling conventional and first-principles approaches, and it directly allowed us to predict the domains of thermodynamic predominance for rapid identification of electrolytes on Al–Gd alloys in a given aqueous system.
-
2.
XRD patterns did not show additional phase formation in mixed alloy chemistries except the reduction of Al(111) peak intensity, revealing the loss of crystallinity in the chosen composition range.
-
3.
From the SEM images of the as-deposited film, three distinct regions were identified: large grains typical of fcc phase in Al-rich region, disappearance of these grains in the middle of the compositional spread, and evolution of upright needle like morphology in alloys of high Gd concentrations.
-
4.
Composition-dependent changes in the film formation factor and relative permittivity, and inverse capacitance were estimated indicating the influence of Gd addition while maintaining the passivation behaviour of alumina. These changes were linear and could be directly correlated to the nanoscopic change of the morphology as a function of alloy composition.
References
Maier WF, Stöwe K, Sieg S (2007) Combinatorial and high-throughput materials science. Angew Chem Int Ed Engl 46:6016–6067
Hao H, Huang J, Liu P, Xue Y, Wang J, Ren K, Jin Q, Ji J, Greiner A, Agarwal S (2021) Rapid build-up of high-throughput screening microarrays with biochemistry gradients via light-induced thiol–ene “click” chemistry. J Mater Chem B 9:3032–3037
Miracle D, Majumdar B, Wertz K, Gorsse S (2017) New strategies and tests to accelerate discovery and development of multi-principal element structural alloys. Scr Mater 127:195–200
Singh A, Talekar M, Tran TH, Samanta A, Sundaram R, Amiji M (2014) Combinatorial approach in the design of multifunctional polymeric nano-delivery systems for cancer therapy. J Mater Chem B 2:8069–8084
Coury FG, Clarke KD, Kiminami CS, Kaufman MJ, Clarke AJ (2018) High throughput discovery and design of strong multicomponent metallic solid solutions. Sci Rep 8:8600
Klemm SO, Schauer J-C, Schuhmacher B, Hassel AW (2011) A microelectrochemical scanning flow cell with downstream analytics. Electrochim Acta 56:4315–4321
Akbarzadeh AR, Ozoliņš V, Wolverton C (2007) First-principles determination of multicomponent hydride phase diagrams: application to the Li-Mg-N-H system. Adv Mater 19:3233–3239
Jain A, Hautier G, Ong SP, Moore CJ, Fischer CC, Persson KA, Ceder G (2011) Formation enthalpies by mixing GGA and GGA + U calculations. Phys Rev B 84:045115
Kirklin S, Meredig B, Wolverton C (2013) High-throughput computational screening of new Li-Ion battery anode materials. Adv Energy Mater 3:252–262
Hattrick-Simpers JR, Tan Z, Oguchi H, Chiu C, Heilweil EJ, Maslar JE, Bendersky LA (2011) A combinatorial characterization scheme for high-throughput investigations of hydrogen storage materials. Sci Technol Adv Mater 12:054207
Hafiz H, Khair AI, Choi H, Mueen A, Bansil A, Eidenbenz S, Wills J, Zhu JX, Balatsky AV, Ahmed T (2018) A high-throughput data analysis and materials discovery tool for strongly correlated materials. npj Comput Mater 4:63
Sanvito S, Oses C, Xue J, Tiwari A, Zic M, Archer T, Tozman P, Venkatesan M, Coey M, Curtarolo S (2017) Accelerated discovery of new magnets in the Heusler alloy family. Sci Adv 3:e1602241
Mardare AI, Ludwig A, Savan A, Hassel AW (2014) Electrochemistry on binary valve metal combinatorial libraries: niobium-tantalum thin films. Electrochim Acta 140:366–375
Iwasaki Y, Ishida M, Shirane M (2020) Predicting material properties by integrating high-throughput experiments, high-throughput ab-initio calculations, and machine learning. Sci Technol Adv Mater 21:25–28
Hautier G, Jain A, Ong PS (2012) From the computer to the laboratory: materials discovery and design using first-principles calculations. J Mater Sci 47:7317–7340
Mao Z, Seidman DN, Wolverton C (2011) First-principles phase stability, magnetic properties and solubility in aluminum – rare-earth (Al – RE) alloys and compounds. Acta Mater 59:3659–3666
Petit L, Svane A, Szotek Z, Temmerman WM (2005) First-principles study of rare-earth oxides. Phys Rev B: Condens Matter Mater Phys 72:205118
Potyrailo R, Rajan K, Stoewe K, Chisholm B, Dakota N, States U, Lam H (2011) Combinatorial and high-throughput screening of materials libraries : review of state of the art. ACS Comb Sci 13:579–633
Mardare AI, Yadav AP, Wieck AD, Stratmann M, Hassel AW (2008) Combinatorial electrochemistry on Al–Fe alloys. Sci Technol Adv Mater 9:035009
Blazey KW, Rohrer H (1968) Antiferromagnetism and the magnetic phase diagram of GdAlO3. Phys Rev 173:574–580
Cashion JD, Cooke AH, Thorp TL, Wells MR (1970) Magnetic properties of gadolinium ortho-aluminate. Proc R Soc A 318:473–495
Petrov D (2011) Nanocrystalline GdAlO3: XPS, EPR and magnetic susceptibility studies. Appl Phys A: Solids Surf 104:1237–1242
Kukli K, Hatanpää T, Ritala M, Leskelä M (2007) Atomic layer deposition of gadolinium oxide films. Chem Vap Deposition 13:546–552
Cheng X, Xu D, Song Z, He D, Yu Y, Zhao Q, Shen D (2009) Characterization of gadolinium oxide film by pulse laser deposition. Appl Surf Sci 256:921–923
Dakhel AA (2005) Optical and dielectric properties of gadolinium–erbium oxide films prepared on Si(100) substrate. Appl Phys A: Solids Surf 80:1033–1037
Sahoo NK, Senthilkumar M, Thakur S, Bhattacharyya D (2002) Correlation of optical and microstructural properties of Gd2O3 thin films through phase-modulated ellipsometry and multi-mode atomic force microscopy. Appl Surf Sci 200:219–230
Hong M, Kwo J, Kortan AR, Mannaerts JP, Sergent AM (1999) Epitaxial cubic gadolinium oxide as a dielectric for gallium arsenide passivation. Science 283:1897–1900
Zhou J-P, Chai C-L, Yang S-Y, Liu Z-K, Song S-L, Li Y-L, Chen N-F (2004) Properties of high k gate dielectric gadolinium oxide deposited on Si (1 0 0) by dual ion beam deposition (DIBD). J Cryst Growth 270:21–29
Watanabe K, Sakairi M, Takahashi H, Takahiro K, Nagata S, Hirai S (2001) Formation of composite oxide films on aluminum by sol-gel coating and anodizing —for the development of high-performance aluminum electrolytic capacitors. Electrochemistry 69:407–413
Habazaki H, Koyama S, Aoki Y, Sakaguchi N, Nagata S (2011) Enhanced capacitance of composite anodic ZrO2 films comprising high permittivity oxide nanocrystals and highly resistive amorphous oxide matrix. ACS Appl Mater Interfaces 3:2665–2670
Woldemedhin MT, Raabe D, Hassel AW (2011) Grain boundary electrochemistry of Β-Type Nb–Ti alloy using a scanning droplet cell. Phys Status Solidi Appl Mater Sci (a) 208:1246–1251
Hassel AW, Lohrengel MM (1997) The scanning droplet cell and its application to structured nanometer oxide films on aluminium. Electrochim Acta 42:3327–3333
Small L, Cook A, Apblett C, Ihlefeld JF, Brennecka G, Duquette D (2012) An automated electrochemical probe for evaluation of thin films. J Electrochem Soc 159:F87–F90
Casillas N, Charlebois SJ, Smyrl WH, White HS (1993) Scanning electrochemical microscopy of precursor sites for pitting corrosion on titanium. J Electrochem Soc 140:L142–L145
Woldemedhin MT, Raabe D, Hassel AW (2012) Characterization of thin anodic oxides of Ti–Nb alloys by electrochemical impedance spectroscopy. Electrochim Acta 82:324–332
Kollender JP, Mardare AI, Hassel AW (2013) Photoelectrochemical scanning droplet cell microscopy (PE-SDCM). ChemPhysChem 14:560–567
Hafner M, Mardare AI, Hassel AW (2013) Vapour phase co-deposition of Al–Cu thin film alloys. Phys Status Solidi A 210:1006–1012
Hassel AW, Fushimi K, Seo M (1999) An agar-based silver|silver chloride reference electrode for use in micro-electrochemistry. Electrochem Commun 1:180–183
Klemm SO, Schauer J-C, Schuhmacher B, Hassel AW (2011) High throughput electrochemical screening and dissolution monitoring of Mg – Zn material libraries. Electrochim Acta 56:9627–9636
Pourbaix M (1974) Atlas of electrochemical equilibria in aqueous solutions. National Association of Corrosion Engineers, Houston, USA
Bard AJ, Parsons R, Jordan J (1985) Standard potentials in aqueous solution. Taylor & Francis, London, UK
Geochemist's Workbench (2019). https://www.gwb.com. Accessed 19 Jul 2019
FactSage (2019). http://www.factsage.com. Accessed 05 May 2019
Medusa (2019). https://www.kth.se/che/medusa. Accessed 18 Nov 2019
Materials Project (2020). https://materialsproject.org. Accessed 23 Apr 2020
Jain A, Hautier G, Moore CJ, Ong SP, Fischer CC, Mueller T, Persson KA, Ceder G (2011) A high-throughput infrastructure for density functional theory calculations. Comput Mater Sci 50:2295–2310
Persson KA, Waldwick B, Lazic P, Ceder G (2012) Prediction of solid-aqueous equilibria: scheme to combine first-principles calculations of solids with experimental aqueous states. Phys Rev B: Condens Matter Mater Phys 85:235438
Car R, Parrinello M (1985) Unified approach for molecular dynamics and density-functional theory. Phys Rev Lett 55:2471–2474
Gschneidner KA, Calderwood FW (1988) The Al-RE (aluminum-rare earth) systems. Bull Alloy Phase Diagr 9:658–668
Mardare AI, Grill CD, Pötzelberger I, Etzelstorfer T, Stangl J, Hassel AW (2016) Anodic oxide formation on aluminium-terbium alloys. J Solid State Electrochem 20:1673–1681
Revie RW (2000) Uhlig’s corrosion handbook. John Wiley & Sons, Inc., New York, USA
Schweitzer GK, Pesterfield LL (2010) The aqueous chemistry of the elements. Oxford University Press, Oxford, UK
Crossland AC, Thompson GE, Skeldon P, Wood GC, Smith CJE, Habazaki H, Shimizu K (1998) Anodic oxidation of Al-Ce alloys and inhibitive behaviour of cerium species. Corros Sci 40:871–885
Herrera-Erazo AE, Habazaki H, Shimizu K, Skeldon P, Thompson GE (2000) Anodic film growth on Al–Nd alloys. Corros Sci 42:1823–1830
Habazaki H, Skeldon P, Thompson GE, Wood GC, Shimizu K (1997) Anodic film formation on a sputter-deposited amorphous Al–40 At. % Sm alloy. J Mater Res 12:1885–1891
Torrescano-Alvarez JM, Curioni M, Habazaki H, Hashimoto T, Skeldon P, Zhou X (2019) Incorporation of alloying elements into porous anodic films on aluminium alloys: the role of cell diameter. Electrochim Acta 296:783–789
Thompson GE, Habazaki H, Shimizu K, Sakairi M, Skeldon P, Zhou X, Wood GC (1999) Anodizing of aluminium alloys. Aircr Eng Aerosp Technol 71:228–238
Randall JJ, Bernard WJ (1975) A radiotracer study of the anodization of aluminum in aqueous phosphate solutions. Electrochim Acta 20:653–661
Takahashi H, Fujimoto K, Nagayama M (1988) Effect of pH on the distribution of anions in anodic oxide films formed on aluminum in phosphate solutions. J Electrochem Soc 135:1349–1353
Firsching FH, Brune SN (1991) Solubility products of the trivalent rare-earth phosphates. J Chem Eng Data 36:93–95
Liu X, Byrne RH (1997) Rare earth and yttrium phosphate solubilities in aqueous solution. Geochim Cosmochim Acta 61:1625–1633
Byrne RH, Kim K-H (1993) Rare earth precipitation and coprecipitation behavior: the limiting role of PO43− on dissolved rare earth concentrations in seawater. Geochim Cosmochim Acta 57:519–526
Cetiner ZS, Wood SA, Gammons CH (2005) The aqueous geochemistry of the rare earth elements. Part XIV. The solubility of rare earth element phosphates from 23 to 150 °C. Chem Geol 217:147–169
Shahzad K, Mardare CC, Recktenwald D, Mardare AI, Hassel AW (2019) Formation of nano-scale composite anodic films on aluminium-holmium alloys. Electrochim Acta 297:888–904
Zhou F, LeClere DJ, Garcia-Vergara SJ, Hashimoto T, Molchan IS, Habazaki H, Skeldon P, Thompson GE (2010) Incorporation and migration of phosphorus species in anodic alumina films containing tungsten tracer layers. J Electrochem Soc 157:C437
Orazem ME, Frateur I, Tribollet B, Vivier V, Marcelin S, Pebere N, Bunge AL, White EA, Riemer DP, Musiani M (2013) Dielectric properties of materials showing constant-phase-element (CPE) impedance response. J Electrochem Soc 160:C215–C225
Hsu CH, Mansfeld F (2001) Technical note: concerning the conversion of the constant phase element parameter Y0 into a capacitance. Corrosion 57:747–748
Brug GJ, van den Eeden ALG, Sluyters-Rehbach M, Sluyters JH (1984) The analysis of electrode impedances complicated by the presence of a constant phase element. J Electroanal Chem Interfacial Electrochem 176:275–295
Hirschorn B, Orazem ME, Tribollet B, Vivier V, Frateur I, Musiani M (2010) Determination of effective capacitance and film thickness from constant-phase-element parameters. Electrochim Acta 55:6218–6227
Hirschorn B (2010) Distributed time-constant impedance responses interpreted in terms of physically meaningful properties. Doctoral dissertation, University of Florida, Gainesville
Hirschorn B, Orazem ME, Tribollet B, Vivier V, Frateur I, Musiani M (2010) Constant-phase-element behavior caused by resistivity distributions in films I. Theory. J Electrochem Soc 157:C452–C457
Funding
Open access funding provided by Johannes Kepler University Linz. The financial support by the Austrian Federal Ministry of Science, Research and Economy and the National Foundation for Research, Technology and Development through funding of the Christian Doppler Laboratory for Combinatorial Oxide Chemistry (COMBOX) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shahzad, K., Mardare, C.C., Mardare, A.I. et al. Mixed oxide growth on combinatorial aluminium–gadolinium alloys — a thermodynamic and first-principles approach. J Solid State Electrochem 25, 2479–2493 (2021). https://doi.org/10.1007/s10008-021-05012-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-021-05012-x