
Abstract
The DFT calculations at the B3LYP level with 6-311G** basis set were carried out in order to reveal whether tautomerization or decarboxylation is responsible for the instability of 2,2-di(pyridin-2-yl)acetic (DPA) and 1,8-diazafluorene-9-carboxylic (DAF) acids. The carboxyl protons in both compounds are involved in the intramolecular hydrogen bonds (the pyridine nitrogen atoms are the hydrogen bond acceptors). Although formation of two intramolecular OH···N hydrogen bonds in the enols of both carboxylic acids enables effective electron delocalization within the quasi rings (···HO − C = C − C = N), only ene-1,1-diol of DAF has somewhat lower energy than DAF itself (ΔE is ca. 7 kcal mol-1). DPA and its enediol have comparable energies. Migration of the methine proton toward the carbonyl oxygen atom (to form enediols) requires overstepping the energy barriers of 55-57 kcal mol-1 for both DPA and DAF. The enaminone tautomers of the acids, formed by migration of this proton toward the pyridine nitrogen atom, are thermodynamically somewhat more stable than the respective enediols. The energy barriers of these processes are equal to ca. 44 and 62 kcal mol-1 for DPA and DAF, respectively. Thus, such tautomerization of the acids is not likely to proceed. On the other hand, the distinct energetic effects (ca. 15 kcal mol-1) favor decarboxylation. This process involves formation of (E)-2-(pyridin-2(1H)-ylidenemethyl)pyridine and its cyclic analogue followed by their tautomerization to (dipyridin-2-yl)methane and 1,8-diazafluorene, respectively. Although the later compound was found to be somewhat thermodynamically more stable, kinetic control of tautomerization of the former is more distinct.
Similar content being viewed by others

Introduction
Ene-1,1-diols, often called enols of carboxylic acids, are intermediates in hydration of ketenes [1, 2]: \( {{\hbox{R}}_2}{\hbox{C}} = {\hbox{C}} = {\hbox{O}} + {{\hbox{H}}_2}{\hbox{O}} \to {{\hbox{R}}_2}{\hbox{C}} = {\hbox{C}}{\left( {\hbox{OH}} \right)_2} \to {{\hbox{R}}_2}{\hbox{CH}} - {\hbox{C}}{{\hbox{O}}_2}{\hbox{H}} \). These compounds are less stable then the corresponding carboxylic acids being energetically favored by delocalization of the lone electron pair of the hydroxy oxygen atom (Fig. 1) [1]. It is known, however, that ene-1,1-diols that contain bulky aromatic groups in the molecule, are more stable [1–4]. The hindered protonation of the β carbon atom in \( {\hbox{A}}{{\hbox{r}}_2}{{\hbox{C}}^\beta } = {\hbox{C}}{({\hbox{OH}})_2} \) caused by ortho alkyl groups (Ar = 2,4,6-trimethylphenyl, 2,4,6-triisopropylphenyl or 2,3,4,5,6-pentamethylphenyl) is responsible for such a behavior. Enediol of cyclopentadiene-1-carboxylic acid (Fig. 2) was found to be relatively stable [5]. Two intramolecular hydrogen bonds of the resonance-assisted hydrogen bond (RAHB) type [6–9] are expected to be present in its derivative annulated with two pyridine rings (Fig. 3). Unfortunately, neither 1,8-diazafluorene-9-carboxylic (9H-cyclopenta[1,2-b;3,4-b′]dipyridine-9-carboxylic) acid nor its enol are known, but close analogue of the former compound, 2,2-di(pyridin-2-yl)acetic acid, was claimed to be obtained earlier from 2-diazo-1,2-di(pyridin-2-yl)ethanone [10]. Since numerous heterocyclic acetic acids are potentially interesting as anti-arthritic agents [11, 12], the compounds mentioned above seem worthy to be studied from point of view of their stability.

Relation between carboxylic acids and ene-1,1-diols

Cyclopentadiene-1-carboxylic acid and its enol

Relation between 1,8-diazafluorene-9-carboxylic acid and its enol
Loosing of the CO2 molecule by 2- and 4-pyridylacetic acids proceeds smoothly even below 100 °C (this process is much more difficult for 3-pyridylacetic acid) [11–17]. 2-(Pyridin-2- and 4-yl)phenylacetic acids are also exceptionally unstable: benzylpyridines were found to be the only products of the acid hydrolysis of 2-(pyridin-2- and 4-yl)phenylacetonitriles [18]. On the other hand, the enediol and enaminone tautomeric forms of the closely related 2,2-di(pyridin-2-yl)acetic acid (Fig. 4) can be stabilized by two intramolecular hydrogen bonds. Its tendency to decarboxylate is not reported in the only paper devoted to synthesis and properties of this compound [10].

Relation between 2,2-di(pyridin-2-yl)acetic (R = H) and and 1,8-diazafluorene-9-carboxylic (R = none) acids and its tautomers
Decarboxylation of carboxylic acid involves formation of carbon dioxide and an organic residue [16]. Organic product is usually stabilized by delocalization of its unshared electron pair. Formation of the intermediate zwitterion \( {\hbox{HOCO}} - {{\hbox{C}}_5}{{\hbox{H}}_4}{\hbox{N}}{ \to^\odot }{\hbox{OCO}} - {{\hbox{C}}_5}{{\hbox{H}}_4}{{\hbox{N}}^\oplus }{\hbox{H}} \) is a common step in the mechanism of enzyme-catalyzed decarboxylations of 2- and 4-pyridylacetic acids [12, 14, 17]. Electron delocalization is particularly important in enzymatic reactions, where an “electron sink” is generally provided by a coenzyme [16].
Despite their instability, the labile compounds may appear as intermediates in numerous (bio)chemical processes. Explanation what are the products these derivatives are transformed to, seems very important. Numerous attempts done in our laboratory to obtain 2,2-di(pyridin-2-yl)acetic acid (1a, Fig. 5) from 2-diazo-1,2-di(pyridin-2-yl)ethanone according to the known procedure [10] as well as by hydrolysis of 2,2-di(pyridin-2-yl)acetonitrile (the standard procedure) were unsuccessful. Irrespective of the reason of inaccessibility of this compound to us, its properties can still be discovered by quantum chemical calculations. In the present paper 2,2-di(pyridin-2-yl)acetic (1a) and 1,8-diazafluorene-9-carboxylic (1b) acids are considered to be susceptible to tautomerization and decarboxylation. The related pyridylacetic acids were used earlier as the models when studying decarboxylation mechanism of aminoacids [19]. It is noteworthy that numerous heterocyclic derivatives of acetic acid are potentially interesting as anti-arthritic agents [20].

Formulas of the compounds studied and the numbering of the heavy atoms
Computational details
Density functional theory (DFT, see, e.g., [21]) has been recognized for years to be capable of predicting the very accurate conformations of the covalently bonded systems. Thus, its use in the present paper is justifiable. All calculations were carried out using B3LYP hybrid functional [22, 23] with the 6-311G** basis set [24].
Formulas of 2,2-di(pyridin-2-yl)acetic (1a) and 1,8-diazafluorene-9-carboxylic (1b) acids, their enol (2a,b) and enaminone (3a,b) tautomeric forms as well as the (expected) intermediates (4a,b) and final products of the decarboxylation process (5a,b) are depicted in Fig. 5, which also shows numbering of heavy atoms. It should be noted that the carboxyl hydrogen atom H9 (in 1, 2 and 3) has the same number even if it is bound to N1 (in 4) or C7 (in 5). Formulas of the transition states that are (or may be) formed during decarboxylation of 1a and 1b can be seen in Fig. 6. The geometry optimization of all substrates, products and transition states was carried out first. Then, the harmonic frequencies were calculated to establish the types of stationary points, and to find the zero-point energy (ZPE) corrections for the energy barriers and the energetic effects of the processes investigated in the present work. All frequencies for systems 1–5(a,b) were real. On the other hand, one imaginary frequency was found for transition states TS(1-4)a,b. Analysis of the eigenvectors corresponding to the imaginary frequencies provided us with the information about the possible products ”on each side” of a transition state at a given stage of the reaction.

Formulas of the transition states studied in this work
Geometry optimization for the transition state is much more complex than for the stable system. This prompted us to carry out some initial, point-wise calculations, corresponding to the so-called distinguished reaction coordinate (DRC). This procedure is not recommended for the accurate description of the reaction path, for example on account of possible discontinuities of energy (and geometrical parameters) as a function of DRC (i.e., some selected geometrical parameter, e.g., bond length). It may, however, provide a reasonable initial structure for which typical algorithms are capable of converging quickly to the required transition state. Therefore, the C7C8 bond was selected as the DRC in reactions 1 → 4 + CO2. Its length, R, was gradually increased from the equilibrium value found for 1 (the applied increment was 0.1 Å), the remaining parameters were optimized, and the geometry corresponding to the highest energy was determined. Since H9 turned out to still be bound to the carboxylic oxygen atom, it was placed in the middle of the O9...N1 distance prior to running the transition state search. On the other hand, the two internal coordinates r 1 = H9N1 and r 2 = H9C7 (cf. Fig. 5) were selected for the description of the proton migration in the 4 → 5 tautomerization. Energies of structures optimized for the remaining geometrical parameters were then computed at a grid of selected linearly independent points. Then, the cross-section of PES (see Fig. 7 for an example), approximated by the 3 rd degree polynomial, was subjected to straightforward search of the saddle point. All initial structures obtained in this way turned out to converge quickly to the corresponding transition states by following (maximizing along) the lowest (negative) Hessian eigenmode. In order to follow the reaction paths, concept of the so-called intrinsic reaction coordinate (IRC) [25] in mass-weighted Cartesian coordinates was used. All calculations were carried out with the parallel version of the PQS quantum chemistry package [26, 27].

The cross-section of the potential energy surface (PES) corresponding to the proton migration in 4a → 5a reaction (see text for the definition of the internal coordinates)
Results and discussion
Molecular geometries
The optimized structures of carboxylic acids as well as these of the enol and enaminone tautomeric forms and products of their decarboxylation (dipyridyl-2-ylmethane and its 1,2-dihydro tautomeric form) are depicted in Fig. 8. Their most important geometrical parameters, as well as geometrical parameters of the transition states TS(1-4)a,b are presented in Tables 1 and 2.

Optimized structures of the substrates and products of the tautomerization/decarboxylation processes
The most significant conclusions regarding the molecular geometries are as follow:
-
1.
Arrangement of the N1 and N1′ atoms in the most stable conformer of 2,2-di(pyridin-2-yl)acetic acid 1a is trans-like relative to the C2C7C2′ link (Fig. 8). Turning of one pyridine ring in 1a by ca. 90° around the C7C2′ bond increases the energy by only 0.3 kcal mol-1. Linking of the pyridine rings via the C3C3′ bond in 1b prevents their free rotation and makes the molecule rigid. Note that the N1C2C7C2′ and N1′C2′C7C2 torsion angles in 1b are close to 180°, i.e., the pyridine rings in the molecule are nearly coplanar. The hydroxyl groups both in 1a and 1b are involved in the intramolecular hydrogen bond with the N1 atom being the hydrogen bond acceptor. The OH bond in 1a is somewhat longer than 0.99 Å (a typical OH bond length in the dimers of ordinary carboxylic acids where very strong intermolecular hydrogen bonds are present). On the other hand, the OH bond in 1b is shorter (ca. 0.98 Å). Rigidity of the 1b molecule and, consequently, steeper energy increase along the interring torsional motions, prevent the H9 and N1 atoms to approach each other as close as in 1a. This results in elongation (weakening) of the H···N1 hydrogen bond (1.918 Å in 1b vs. 1.738 Å in 1a) and shortening (strengthening) of the O9H bond. The carbon-nitrogen bond lengths in the pyridine rings of 1a and 1b are close to these in pyridine itself (1.337 Å; geometry optimized at the B3LYP/6-311G** level). The same applies to the ring carbon-carbon bond lengths which are comparable to the average bond length in pyridine (1.393 Å). Slight elongation of the carbon-nitrogen bonds in 1a and 1b is observed for the ring involved in the hydrogen bonding. The valence CNC angles in these systems are also comparable to those found in the molecule of pyridine (117.2°).
-
2.
Formation of the H···N1 and H···N1′ hydrogen bonds in 2a and 2b affects the hybridization of C7 and shortens the C7C8 bond lengths (1.419 Å in 2a and 1.376 Å in 2b) as compared to these in 1a and 1b. These bonds resemble more the carbon-carbon bonds in the benzene ring (∼1.39 Å) rather than those in alkenes (∼1.33 Å). It should be noted that C7C8 bond in 2a is longer, while this in 2b is shorter than 1.39 Å. Effective electron delocalization in the quasi rings that involve OH···N fragments results also in shortening of the C7C2 and C7C2′ bonds both in 2a and 2b (they are still significantly longer than the aromatic carbon-carbon bonds) as compared to these in 1a and 1b (the C7C8, C7C2 and C7C2′ bonds in their molecules are typical carbon-carbon single bonds). The H9O9 and the hydrogen H···N1(′) bond lengths in 2a,b follow the same pattern as these in the molecules of 1a,b. Moreover, C3C3′ bond in 2b is much longer than C7C2(′) and C2(′)C3(′) bonds (which have comparable lengths, cf. Table 1). Thus, aromaticity of the five membered ring in 2b is doubtful what negates stabilization of this form by resonance suggested in Fig. 3.
-
3.
The NH···O hydrogen bonds are present in the enaminone tautomeric forms 3a and 3b (these bonds are longer than the OH···N hydrogen bonds in 2a and 2b). Contribution of the zwitterionic structures of 3 (cf. Figure 5) is manifested in shortening of the C7C8 bonds (this effect is not as significant as for 2) and elongation of the C8O10 bonds as compared to 1.
-
4.
4a and 4b seem likely to be the intermediate products of decarboxylation of 1a and 1b, respectively. Both these molecules are planar. Hydrogen atom involved in the H···N1 hydrogen bonding in 1a and 1b is bound to N1. The N1H bond lengths (1.009 Å in both systems) are in between these found for pyrrole (1.006 Å) and aliphatic amines (1.015 Å; geometry optimized at the B3LYP/6-311G** level). The C7C2 and C7C2′ bond lengths are intermediate between these of the single and double carbon-carbon bonds. In both 4a and 4b the C7C2 bonds are shorter, while C7C2′ are longer than 1.39 Å (Table 1).
-
5.
5a and 5b are probably the final products of decarboxylation of 1a and 1b. Their geometrical parameters remain similar to these found for 1a and 1b, as well as those in pyridine itself. Note that the pyridine rings in 5b are coplanar.
Reaction paths and energetic effects
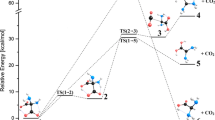
Calculations of the energetic effects of the most intuitive reactions 1 → 5 + CO2 reveal that products are by ca. 13.6 (case a) and ca. 15.8 kcal mol-1 (case b) more stable than substrates. All energetic effects |Eproducts - Esubstrates| and energy barriers ETS - Esubstrates/products discussed (denoted as ΔE) were corrected for the zero-point energy, ZPE. They are depicted in Fig. 9. The data used in order to obtain these values (i.e., total energies of the molecules and ZPE corrections of all stable structures and transition states) are reported in Table 3. Note that for the brevity reason the ΔG values were calculated for decarboxylation processes only. Comparable entropies of the substrates and products for the tautomerization processes suggest that the ΔE and ΔG values also approximate each other.

Reaction paths, ZPE corrected energy barriers and energetic effects (ΔE in kcal mol-1) in tautomerization and decarboxylation of 1a and 1b. The calculated ΔG values (for decarboxylation processes only) are reported in parentheses
Ene-1,1-diols 2 have lower energies than the respective acids 1: the energy lowering is equal to approximately 0.7 and 7 kcal mol-1 for 2a and 2b, respectively (Fig. 9). The energy barriers for the proton migration from C7 toward the carbonyl oxygen atom O10 (this processes proceed via TS1 transition states) are nearly the same for a and b and amount to ca. 55 kcal mol-1. The enaminone tautomeric forms 3a,b are thermodynamically somewhat more stable than 2a,b (by nearly 4 kcal mol-1 in the case of 3b). However, the energy barriers for the migration of proton H7 toward N1′, associated with the presence of TS2a and TS2b transition states, differ significantly: the calculated ΔE values are equal to ca. 44 and ca. 62 kcal mol-1 for 1a → 3a and 1b → 3b, respectively. In a view of finding more energetically favorable processes from the thermodynamic and kinetic point of view (vide infra) we conclude that tautomerization of 1a and 1b is not likely to proceed. Among all investigated processes, namely 1 → 2, 1 → 3, and 1 → 5 + CO2, the most distinct energetic effect can be seen for 1 → 5 + CO2 (for both a and b, cf. Fig. 9). These processes will be discussed in detail.
Rupture of the C7C8 bond in the 1 → 4 + CO2 reaction is probably followed by migration of the carboxylic hydrogen atom to C7 (this results in formation of 5). The concerted and step-wise mechanisms have to be considered here. In the first one, the eigenmode (atomic displacements) corresponding to the imaginary frequency should correspond to simultaneous detachment of CO2 and migration of the carboxylic proton H9 toward C7. The later mechanism is expected to involve a few different transition states. Case a will be considered first. A quick glance at the structure of 1a points at the second, i.e., step-wise mechanism. The reason for this is a significant energy increase when H9 approaches C7, while C7 is still bound to C8. It is shown in Fig. 10 as a steep, violet (turning to blue at larger values of R) “slope” on a cross-section of the potential energy hypersurface of 1a. In addition, elongation of the C7C8 bond reveals that there is a clear trend for the H9 atom to approach N1, i.e., to form 4a. Therefore one should search for a transition state that appears in the course of reaction 1a → TS3a → 4a + CO2. It was found to be nearly 20 kcal mol-1 above 1a. The calculated ΔG value corresponding to the reported ΔE amounts to 18.33 kcal mol-1. This value is known to be low enough for the process that is fast under standard conditions. Analysis of geometrical parameters shows that C7C8 bond in TS3a is significantly longer than that in 1a, and that the H···N1 bond remains close to that of 4a (cf. Tables 1 and 2). The structure of TS3a is depicted in Fig. 11 (the eigenmode, corresponding to the imaginary frequency equal to i320cm-1, is also shown there). Following the reaction path from TS3a along the intrinsic reaction coordinate (IRC), 1a or 4a + CO2 were obtained. No energy lowering is observed at this stage of decarboxylation. However, value of the Gibbs free energy of the products calculated at 298 K is by more than 12 kcal mol-1 lower than that of 1a. Although rotational and vibrational entropies were also changed (ΔSrot = 11.9 cal mol-1 K-1 and ΔSvib = -12.2 cal mol-1 K-1), significant increase in the translational entropy (ΔStrans = 36.6 cal mol-1 K-1) gives the major contribution to the reported value of ΔG. The low energy barrier being < 20 kcal mol-1 (corresponding to energy of quanta in the near IR region) as well as negative value of ΔG clearly show that 1a decomposes spontaneously (and rapidly) to form 4a and CO2. This proves that Eistert and Schade [10] could not obtain 1a. It is probably why we were not able to obtain this compound starting from 2-diazo-1,2-di(pyridin-2-yl)ethanone [10] as well as by hydrolysis of 2,2-di(pyridin-2-yl)acetonitrile (the standard procedure).

Cross-section of the potential energy surface (PES) of 1a corresponding to the rupture of C7C8 (variable R) bond and simultaneous migration of the hydroxyl proton toward C7 (variable r 2)

Transition states TS3a and TS4a. The arrows show the atomic displacement corresponding to the imaginary frequencies
4a is probably an intermediate in the process of decarboxylation of 1a. In order to obtain 5a, which is the most stable system studied (at least at the B3LYP level, cf. Fig. 9 and Table 3), the C7H9 bond has to be formed and, at the same time, the N1H9 bond has to be broken. It was not clear to us which is the mechanism of this step (i.e., unimolecular or bimolecular). It was initially assumed that the rearrangement takes place within the single molecule. This process is accompanied by breaking of the planar symmetry of 4a (note that C7 atom has to change its hybridization from sp2 in 4a to sp3 in 5a). The search for another transition state (TS4a) was carried out in the way described in Sect. 2. Its structure along with the imaginary eigenmode (i1834cm-1) is shown in Fig. 11. Location of H9 more or less half way between N1 and C7 (cf. Table 2) is the characteristic feature of this transition state. The energy barrier ΔE = 42.37 kcal mol-1 that has to be overstepped, corresponds to ΔG value of ca. 38 kcal mol-1. It is two times higher than that for the CO2 detachment. This seems to be mostly due to the high strain within the 4-membered ring formed in the TS4a transition state. Thus, this step of the decarboxylation process, though spontaneous from the thermodynamic point of view, is expected to be much slower than the former one.
An alternative mechanism involves two molecules of 4a. The B3LYP calculations show, that they are capable of forming a relatively stable dimer possessing the C2 symmetry. It is depicted on Fig. 12a; the binding energy, counterpoise (CP) corrected for the basis set superposition error [28] and ZPE corrected, is equal to 4.26 kcal mol-1. The transition state, corresponding to the imaginary frequency of i1281 cm-1, was found to be only 14.58 kcal mol-1 above the dimer (the ZPE corrected value; the corresponding ΔG value is 11.41 kcal mol-1). Analysis of the imaginary eigenmode shows that this is the transition state for the migration of the H9 atom from N1 atom of one 4a molecule to the C7 atom of the other 4a molecule (actually, two molecules of 5a were finally obtained by following the reaction path along the intrinsic reaction coordinate). The B3LYP energetic effect of the overall reaction, i.e., (4a)2 → 2 5a, is 10.85 kcal mol-1 per molecule (ΔG = 17.14 kcal mol-1). However, it should be noted that the dimer predicted by B3LYP calculations is stabilized by the interactions of the NH fragment of one molecule (a rather weak dipole) with the non-polar C…C…C system of the other molecule (the quadrupole, cf. Fig. 12a). Thus, the dispersive-type interactions that show the U disp ∼ r -6 dependence on distance, are not negligible as compared to the weak electrostatic dipole-quadrupole interactions U dip-quad ∼ r -4. The same type of interactions are expected in the transition state. Since DFT is known to poorly describe the dispersive interactions, we decided to investigate this problem using the MP2 method with the same basis set. The geometry optimization of the dimer of 4a was carried out at the MP2/6-311G** level, using the B3LYP structure as an initial guess. This time we did not obtain the analogous dimer; two NH···N hydrogen bonds were formed instead. This complex, for which the CP corrected binding energy is 14.29 kcal mol-1 (the MP2 frequencies were not calculated for practical reasons, but the ZPE estimate obtained at the DFT level, would further reduce this value by ca. 0.7 kcal mol-1), is shown in Fig. 12b. Thus it seems, that the DFT calculations overestimate the dispersive interactions when two molecules of 4a approach each other.

The dimers of 4a predicted at (a) B3LYP and (b) MP2 levels
In a view of the MP2 results we conclude that the unimolecular mechanism is responsible for the formation of 5a. Note that the NH···N links are typically responsible for the chemical exchange of the labile protons. Actually, compounds 4a and 5a were obtained by following the reaction path from TS4a along the intrinsic reaction coordinate. It should be noted that the reported energy barrier of ca. 40 kcal mol-1 is comparable to that found for the 1a → TS2a → 3a reaction. However, low energy barrier of the CO2 detachment, and significantly higher stability of 5a as compared to 3a, proves that synthesis of 1a [10] could not be successful: this compound spontaneously decarboxylates when it is formed.
Structures of the TS3b and TS4b transition states in decarboxylation of 1b as well as imaginary eigenmodes (corresponding to frequencies equal to i332cm-1 and i2050cm-1, respectively) are depicted in Fig. 13. Weaker intramolecular hydrogen bond in 1b as compared to that in 1a (cf. Table 1), is to some extent responsible for the lower energy barrier (ΔG ≈ 13 kcal mol-1, cf. Fig. 9) of the dissociation process 1b → 4b + CO2, and for the formation of the N1H bond in 4b. This stage of the reaction is expected to be spontaneous as well: energy lowering (ΔE = 4.8 kcal mol-1, and ΔG = 17.24 kcal mol-1) was observed. The most significant difference between this (b) and previous (a) decarboxylation processes is an increase of the energy barrier associated with the migration of H9 toward C7 in the 4b → 5b tautomerization by more than 20 kcal mol-1. This value refers to both ΔE and ΔG. Note, that we assume the unimolecular mechanism of tautomerization, in spite of finding a relatively stable dimer of 4b at the B3LYP level, analogous to the dimer of 4a, for which the CP corrected binding energy is equal to ca. 6 kcal mol-1 (the imaginary eigenmode for the transition state found for (4b)2 → 2 5b reaction corresponds to i1149cm-1, the ΔE/ΔG energy barriers are ca. 14.7/12.6 kcal mol-1, and the ΔE/ΔG energetic effects are ca. 7.5/13.6 kcal mol-1 per molecule, respectively). However, this time the lengthy MP2 calculations were not carried out. The reason for this can be easily understood when structures involved in the described processes are analyzed. The change in the hybridization of C7 to sp 3 when going from 4b to 5b via TS4b is associated with breaking of the planar symmetry of 4b. Similar behavior is also observed in reaction 4a → 5a. One should keep in mind, however, that the C3C3′ bond makes the molecules 4b and 5b rigid (cf. Sect. 3.1), and this results in a steeper energy increase under torsional distortions. As a consequence, significantly risen energy barrier of the 4b → 5b step (ca. 60 kcal mol-1) may preclude tautomerization to proceed. Thus, 4b may appear to be the final product of decarboxylation of 1b. In addition, it can be seen from Fig. 9 and Table 3 that although 5b is thermodynamically more stable than 5a, kinetic control of the 4a → 5a process is more distinct than that of 4b → 5b.

Transition states TS3b and TS4b. The arrows show the atomic displacement corresponding to the imaginary frequencies
Finally we would like to point out that the bimolecular mechanism was also considered in the case of the enolization process of 1a. The stable dimer, that could initiate the H proton transfer from C7 atom of one molecule of 1a to O10 atom of the other molecule of 1a, was found at the B3LYP level. It is shown in Fig. 14. This time no transition state associated with such a migration was found (the MP2 calculations were not carried out).

The dimer of 1a predicted at B3LYP level
Conclusions
Tautomerization and decarboxylation of 2,2-di(pyridin-2-yl)acetic acid were considered to be responsible for its instability. DFT/B3LYP/6-311G** calculations show that the products of decarboxylation of this acid and its rigid cyclic analogue, 1,8-diazafluorene-9-carboxylic acid, are more energetically favorable than the corresponding ene-1,1-diols (products of migration of the methine proton toward the carbonyl oxygen atom) and enaminones (products of migration of the same proton toward the pyridine nitrogen atom). The later tautomers were found to be thermodynamically more stable than enediols. Aromatization of the five membered ring has no effect on stabilization of ene-1,1-diol of 1,8-diazafluorene-9-carboxylic acid. Detachment of CO2 from 2,2-di(pyridin-2-yl)acetic and 1,8-diazafluorene-9-carboxylic acids to give (E)-2-(pyridin-2(1H)-ylidenemethyl)pyridine and its cyclic analogue, respectively, may be followed by tautomerization of these by-products into (dipyridin-2-yl)methane and 1,8-diazafluorene. The calculated energy barriers for the CO2 detachment correspond to quanta of IR radiation, so they can be easily overstepped in the course of reactions expected to afford 2,2-di(pyridin-2-yl)acetic and 1,8-diazafluorene-9-carboxylic acids. Thus, spontaneous decarboxylation of the former is in line with the unsuccessful synthetic efforts.
Despite their instability, the labile compounds may appear as the intermediates in numerous (bio)chemical processes. Conclusions of the present paper may be valid also for other substituted acetic acids (and their homologues) that carry electronegative heteroatoms in the gamma positions, such as diacylacetic acids and their derivatives (to be also known as unstable compounds).
References
Frey J, Rappoport Z (1996) J Am Chem Soc 118:5169–5181
Allen BM, Hegarty AF, O’Neill P, Nguyen MT (1992) J Chem Soc Perkin Trans 2:927–934
O’Neill P, Hegarty AF (1987) J Chem Soc Chem Commun 744-745
Allen BM, Hegarty AF, O’Neill P (1997) J Chem Soc Perkin Trans 2:2733–2736
Urwyler B, Wirz J (1990) Angew Chem Int Ed Engl 29:790–792
Gilli G, Belluci F, Ferretti V, Bertolasi V (1989) J Am Chem Soc 111:1023–1028
Bertolasi V, Gilli P, Ferretti V, Gilli G (1991) J Am Chem Soc 113:4917–4925
Gilli P, Bertolasi V, Ferretti V, Gilli G (1994) J Am Chem Soc 116:909–915
Bertolasi V, Gilli P, Ferretti V, Gilli G (1996) Chem Eur J 2:925–934
Eistert B, Schade W (1958) Chem Ber 91:1411–1415
Taylor PJ (1972) J Chem Soc Perkin Trans 2:1077–1086
Sicinska D, Lewandowicz A, Vokal B, Paneth P (2001) J Org Chem 66:5534–5536
Katritzky AR, Pozharsky AF (2000) Handbook of Heterocyclic Chemistry, 2nd edn. Pergamon, Elsevier, Amsterdam
von Doering WE, Pasternak VZ (1950) J Am Chem Soc 72:143–147
Sicinska D, Truhlar DG, Paneth P (2001) J Am Chem Soc 123:7683–7686
Headley GW, O’Leary MH (1990) J Am Chem Soc 112:1894–1896
Marlier JF, O’Leary MH (1986) J Am Chem Soc 108:4896–4899
Panizzon L (1944) Helv Chim Acta 27:1748–1756
O’Leary MH (1988) Acc Chem Res 21:450–455, and papers cited therein
British Patents 1,099,389; 1,121,922; 1.139,940; 1,147,068 and 1,164,510.
Parr RG, Yang W (1989) Density-Functional Theory of Atoms and Molecules. Oxford University Press, New York
Becke AD (1993) J Chem Phys 98:5648–5652
Lee C, Yang W, Par RG (1993) Phys Rev B 37:785–769
Krishnan R, Binkley JS, Seeger R, Pople JA (1980) J Chem Phys 72:650–654
Fukui K (1970) J Phys Chem 74:4161–4163
Baker J, Wolinski K, Malagoli M, Kinghorn D, Wolinski P, Magyarfalvi G, Saebo S, Janowski T, Pulay P (2009) J Comput Chem 30(2):317–335
PQS version 3.3, Parallel Quantum Solutions; 2013 Green Acres Road, Fayetteville, AR 72703, USA
Boys SF, Bernardi F (1970) Mol Phys 19:553–566
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Borowski, P., Gawinecki, R., Miłaczewska, A. et al. Instability of 2,2-di(pyridin-2-yl)acetic acid. Tautomerization versus decarboxylation. J Mol Model 17, 857–868 (2011). https://doi.org/10.1007/s00894-010-0780-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-010-0780-y