
Abstract
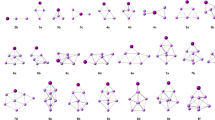
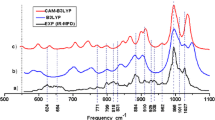
Geometrical structures and relative stabilities of (LiNH2)n (n = 1–5) clusters were studied using density functional theory (DFT) at the B3LYP/6-31G* and B3LYP/6-31++G* levels. The electronic structures, vibrational properties, N–H bond dissociation energies (BDE), thermodynamic properties, bond properties and ionization potentials were analyzed for the most stable isomers. The calculated results show that the Li–N and Li–Li bonds can be formed more easily than those of the Li–H or N–H bonds in the clusters, in which NH2 is bound to the framework of Li atomic clusters with fused rings. The average binding energies for each LiNH2 unit increase gradually from 142 kJ mol−1 up to about 180 kJ mol−1 with increasing n. Natural bond orbital (NBO) analysis suggests that the bonds between Li and NH2 are of strong ionicity. Three-center–two-electron Li–N–Li bonding exists in the (LiNH2)2 dimer. The N–H BDE values indicate that the change in N–H BDE values from the monomer a1 to the singlet-state clusters is small. The N–H bonds in singlet state clusters are stable, while the N–H bonds in triplet clusters dissociate easily. A study of their thermodynamic properties suggests that monomer a1 forms clusters (b1, c1, d2 and e1) easily at low temperature, and clusters with fewer numbers of rings tend to transfer to ones with more rings at low temperature. E g, E HOMO and E av decrease gradually, and become constant. Ring-like (LiNH2)3,4 clusters possess higher ionization energy (VIE) and E g, but lower values of E HOMO. Ring-like (LiNH2)3,4 clusters are more stable than other types. A comparison of structures and spectra between clusters and crystal showed that the NH2 moiety in clusters has a structure and spectral features similar to those of the crystal.

(LiNH2) n (n = 1~5) clusters were studied by the density functional method. The average binding energies for each LiNH2 unit gradually increase from 142 kJ/mol up to about 180 kJ/mol with n increasing. The bonds between Li and NH2 are of strong ionicity.



Similar content being viewed by others

References
Chen P, Xiong ZT, Luo JZ (2002) Nature 420:302–304
Leng HY, Ichikawa T, Hino S (2006) J Power Sources 156:166–170
Hinehliffe A (1977) Chem Phys Lett 45:88–91
Yoshino M, Kcaniya K, Takahashi Y (2005) J Alloys Compd 404–406:185–190
Burk P, Koppel I (1994) Int J Quantum Chem 51:313–318
Velikokhatnyi OI, Kumta PN (2007) Mater Sci Eng B 140:114–122
Tsumuraya T, Shishidou T, Oguchi T (2007) J Alloys Compd 446–447:323–327
Chen YH, Kang L, Zhang CR (2008) Acta Phys Sin 57:4174–4181
Chen YH, Kang L, Zhang CR (2008) Acta Chim Sin 66:2030–2036
Ge GX, Jing Q, Yang Z (2006) Acta Phys Sin 55:4548–4552
Jacobs H, Juza R (1972) Z Anorg Allg Chem 391:271–279
Miwa K, Ohba N, Towata S (2005) Phys Rev B 71:195109
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785–789
Becke AD (1992) J Chem Phys 97:9173–9177
Blanksby SJ, Ellison GB (2003) Acc Chem Res 36:255–263
Frisch MJ, Trucks GW, Schlegel HB et al (2003) Gaussian 03, Revision B.03. Gaussian, Pittsburgh
Douglas BG, Sheridan PM, Jihad IA (2001) J Am Chem Soc 123:5489–5494
Patrick B, Josik P, Georges T (1969) J Mol Struct 4:1–13
Linde G, Juza R (1974) Z Anorg Allg Chem 409:199–214
Philip AC, Paul AA, James WP (2007) J Alloys Compd 446–447:350–354
Bohger JPO, Eßman RR, Jacobs H (1995) J Mol Struct 348:325–328
Joseph WN, George CP (1965) Spectrochim Acta 21:877–882
Zhang CJ, Alavi A (2006) J Phys Chem B 110:7139–7143
Gupta M, Gupta RP (2007) J Alloys Compd 446–447:319–322
Scott AP, Radom L (1996) J Phys Chem 100:16502–16513
Orimo S, Nakamori Y, Eliseo JR (2007) Chem Rev 107:4111–4132
Celotta RJ, Bennett RA, Hall JL (1974) J Phys Chem 60:1740–1745
Acknowledgments
We gratefully acknowledge support from the Key Laboratory for Attapulgite Science and Applied Technology of Jiangsu Province and the Natural Science Foundation of Jiangsu Province. S.Q.Z. thanks the Innovation Project for Postgraduates in Universities of Jiangsu Province (Grant No. CX09B093Z) for partial financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhou, SQ., Zhou, SM., Hu, T. et al. Density functional theory study on (LiNH2)n (n = 1–5) clusters. J Mol Model 17, 235–242 (2011). https://doi.org/10.1007/s00894-010-0717-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-010-0717-5