
Abstract
An agar-degrading archaeon Halococcus sp. 197A was isolated from a solar salt sample. The agarase was purified by hydrophobic column chromatography using a column of TOYOPEARL Phenyl-650 M. The molecular mass of the purified enzyme, designated as Aga-HC, was ~55 kDa on both SDS-PAGE and gel-filtration chromatography. Aga-HC released degradation products in the order of neoagarohexose, neoagarotetraose and small quantity of neoagarobiose, indicating that Aga-HC was a β-type agarase. Aga-HC showed a salt requirement for both stability and activity, being active from 0.3 M NaCl, with maximal activity at 3.5 M NaCl. KCl supported similar activities as NaCl up to 3.5 M, and LiCl up to 2.5 M. These monovalent salts could not be substituted by 3.5 M divalent cations, CaCl2 or MgCl2. The optimal pH was 6.0. Aga-HC was thermophilic, with optimum temperature of 70 °C. Aga-HC retained approximately 90 % of the initial activity after incubation for 1 hour at 65–80 °C, and retained 50 % activity after 1 hour at 95 °C. In the presence of additional 10 mM CaCl2, approximately 17 % remaining activity was detected after 30 min at 100 °C. This is the first report on agarase purified from Archaea.
Similar content being viewed by others

Introduction
Agar, the main cell wall component of red macroalgae, is widely used as a gelling agent of microbiological culture media as well as molecular sieving in DNA electrophoresis and gel-filtration chromatography. Agar produced commercially from the species of genera Gelidium and Gracilaria is composed of agarose and agaropectin (Aoki et al. 1990; Rochas et al. 1986). Agarose consists of 3, 6-anhydro-l-galactose and d-galactose alternately linked by α-1, 3 and β-1, 4 linkages (Kloareg and Quatrano 1988). Agaropectin has the same basic disaccharide-repeating units as agarase with some hydroxyl groups of 3, 6-anhydro-l-galactose residues replaced by sulfoxy or methoxy and pyruvate residues (Hamer et al. 1977).
Agarases are hydrolytic enzymes that degrade agarose into oligosaccharides. They were characterized as either α-agarase (E.C. 3.2.1.158) that cleaves α-1, 3 linkage to produce a series of agaro-oligosaccharides related to agarobiose (Potin et al. 1993) or β-agarase (E.C. 3.2.1.81) that cleaves β-1, 4 linkage to produce neoagarooligosaccharides of series related to neoagarobiose (Kirimura et al. 1999). Agarose-degrading enzymes have been attracting keen interest in many fields of biochemistry and enzymology (Fu and Kim 2010). The neoagarooligosaccharides have various special biological activities, such as inhibition of bacterial growth, slowing down of starch degradation thereby reducing the calorific value of food, and providing anticancer, antivirus, and anti-oxidation activities (Giordano et al. 2006). The β-agarases are also used to recover DNA from agarose gel after electrophoresis (Finkelstein and Rownd 1978; Burmeister and Lehrach 1989).
There have been numerous reports on agarase from isolates belonging to genera of the domain Bacteria, including Acinetobacter, Agarivorans, Alteromonas, Bacillus, Cytophaga, Microbulbifer, Pseudoalteromonas, Pseudomonas, Salegentibacter, Thalassomonas, Vibrio, Zobellia, etc. (Hu et al. 2009; Fu and Kim 2010), many of which are of marine origin. There have been no reports, however, on agarases from halophilic Bacterial strains requiring high (more than 10 %) NaCl concentrations for growth. On the other hand, no reports have been published on agarases from any strains of the domain Archaea, although glycoside hydrolases such as α-amylases (Pérez-Pomares et al. 2003; Hutcheon et al. 2005), chitinase (Zhang et al. 2010), cyclodextrin glycosyltransferase (Bautista et al. 2012), β-xylanase and β-xylosidase (Wainø and Ingvorsen 2003), have been identified from halophilic Archaeal strains, as well as from hyperthermophilic Archaea (Kim and Ishikawa 2010; Rashid et al. 2002; Tanaka et al. 2001).
In the present study, we isolated agarase-producing halophilic archaeal strains belonging to the genus Halococcus from solar salt samples. We describe purification and characterization of the halophilic and thermophilic β-agarase.
Materials and methods
Isolation of agar-degrading extreme halophiles
Agar-degrading extreme halophiles were screened on agar plates of modified JCM medium No. 169, which contained (in per liter) 250.0 g NaCl, 0.75 g casamino acids, 1.0 g yeast extract, 0.3 g trisodium citrate, 2.0 g KCl, 20.0 g MgSO4·7H2O, 0.05 g FeSO4 4H2O, 0.2 g MnSO4 4H2O, 20.0 g Bacto-agar (Difco), pH adjusted to 7.2 with 40 % KOH. Approximately 300 salt samples, domestic and imported, were dissolved in 4 ml of 5 % sterile NaCl solution, spread on the agar plates, and incubated at 37 °C for 2 weeks. Two colonies that formed depression or clearing zones on agar were picked up and purified further by the same plating method.
Tentative identification of isolates
Total DNA was extracted by the method of Cline et al. (1989). The 16S rRNA gene was amplified by PCR with the following forward primer H16S For (5′-CCCTGCGSTCCGSCGT-3′) and reverse primer 23S Rev2 (5′-GCTTATCGCAGCTTGG-3′) (Minegishi et al. 2012). The amplification with Ex Taq DNA polymerase (TaKaRa, Japan) was started by incubation at 96 °C for 5 min, followed by 25 cycles of 20 s at 98 °C, 30 s at 58 °C, and 1 min 30 s at 72 °C, and final extension was done for 2 min at 72 °C. The amplified DNA was cloned by the TA Cloning Kit (Invitrogen) and sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kits (Applied Biosystems) with the following primers; H16S_86F: 5′-GCTCAGTAACACGTGGCCAA-3′ and H16S_351R: 5′-GTAAAGGTTTCGCGCCTGCT-3′ and H16S_314F: 5′-CCGGGCCCTACGGGGCGCAG-3′ and H16S_982F: 5′-GAGAGGAGGTGCATGGCCGC-3′ and H16S_1295R: 5′-CTACCGAATCCAGCTTCATG-3′ on the ABI PRISM 310 Genetic Analyzer (Applied Biosystems).
Enzyme assay
Agarase activity was determined by measuring the increase of reducing sugar released from agarose using the modified dinitro salicyclic acid (DNS) method (Miller 1959). Unless indicated, Agarose-ME (Medium Electroendosmosis, Nacalai Tesque, Kyoto, Japan), with gelling temperature of 35–40 °C, was used as the substrate. The standard assay condition was as follows: reaction mixture containing 0.9 ml of 0.33 % (w/v) Agarose-ME solution in 3.5 M NaCl, 50 mM 2-Morpholinoethanesulfonic acid (MES)-NaOH buffer (pH 6.0) and 0.1 ml of diluted enzyme solution was incubated at 65 °C for 15 min. The reaction mixture was mixed with 1.0 ml of DNS reagent and boiled for 5 min. The mixture was diluted with 4.0 ml of distilled deionized water, and absorbance was read at 535 nm. For the blanks, we placed reaction mixtures containing 0.1 ml of 2.5 M NaCl, 5 mM Tris–HCl buffer without the enzyme. In some experiments, agarose preparations from other suppliers were also used; Agarose-LE (Nacarai Tesque, Japan), Agarose-S (Nippon Gene, Japan), Certified Molecular Biology Agarose (BIO-RAD), Bacto-agar (Difco), and Agar noble (Difco).
Production and purification of the agarase
The agarase-producing Halococcus sp. was cultivated at 37 °C with rotary shaking at 120 rpm in 2.0 liters of a medium HA (in per liter): 250.0 g NaCl, 2.0 g KCl, 20.0 g MgSO4 7H2O, 5.0 g yeast extract, 5.0 g casamino acids, 3.0 g trisodium citrate, 1.0 g sodium glutamate, 36.0 mg FeSO4 4H2O, 0.36 mg MnCl2 4H2O, pH adjusted to 7.2 with 40 % KOH. The cells at late logarithmic phase were collected aseptically by centrifugation at 8000×g for 15 min at 15 °C, then the cell pellet was seeded into 4.0 liters of an induction medium HA-i composed of salt and trace metal components of HA and 0.2 % agarose (Certified Molecular Biology Agarose, Bio-Rad). After incubation for 2–3 days at 37 °C with shaking at 120 rpm, supernatant of the culture obtained by centrifugation was saturated with NaCl, and subjected to hydrophobic column chromatography using a column (2.5 × 15 cm) of TOYOPEARL Phenyl-650 M (TOSOH, Japan) equilibrated with 30 % NaCl, 5 mM Tris–HCl, pH 7.0. After washing with the same buffer, elution was done with the Tris–HCl buffer of step-wise decreasing NaCl concentrations. Fractions with agarase activities were pooled, NaCl added to saturation, loaded to another column of TOYOPEARL Phenyl-650 M (1 × 10 cm), and eluted with a linear decreasing NaCl concentration, from 5.0 M to zero. Agarase-active fractions were collected and dialyzed against 2.5 M NaCl, 5 mM Tris–HCl, pH 7.0, and concentrated with an ultrafiltration device, Amicon Ultra 3 kDa (Millipore). All procedures were done at 4 °C and the relative protein content was estimated by absorbance at 280 nm. Protein concentration of purified enzyme was determined by Lowry method, using bovine serum albumin as the standard.
Characterization of agarase activity
The effect of salt concentrations on the enzyme activity was determined using 0.3 % agarose in the standard buffer (50 mM MES, pH 6.0) containing 0–5.0 M NaCl, 0–3.5 M KCl, or 0–3.5 M LiCl. Divalent cation salts, 0–3.5 M MgCl2 or 0–3.5 M CaCl2 at intervals of 0.5 M.
The optimum pH for the enzyme was examined in the following 50 mM buffers with various pH values: citric acid-NaOH (pH 4.0–6.0), MES-NaOH (pH 5.5–7.0), sodium phosphate (pH 6.5–8.0), glycylglycine-NaOH (pH 7.0–8.5), N-tris (Hydroxymethyl) methyl-4-aminobutanesulfonic acid-NaOH (TABS-NaOH) (pH 8.5–9.5) under the standard assay condition. The effect of temperature was examined under the standard assay condition except that the temperature was varied from 35 to 100 °C.
Thermal stability of the enzyme (~0.04 mg/ml) was determined in the presence of 3.5 M NaCl by incubation at 65, 70, 80, 90 and 95 (in glass test tubes using thermostated water bath) and 100 °C (in 0.2 ml microtubes using heat block of GeneAmp PCR System 9700, Applied Biosystems) for 0–60 min and the residual activities were measured under the standard assay condition. In a separate experiment, stabilities at 100 °C were measured in the absence or presence of 10 mM CaCl2. The enzymatic characterizations were performed three or four times with n = 3 for each samples.
Analysis of hydrolysates of agarose
Agarose-ME was hydrolysed with Aga-HC in the presence of 3.5 M NaCl, 5 mM Tris–HCl, pH 6.0, for 1, 2, 4, 8, and 16 h at 65 °C. The hydrolysates were desalted using ion-exchange resins from Amberlite MB-3 (Organo, Tokyo, Japan) and analyzed by thin-layer chromatography (TLC) on Silica 60 TLC plates (Merck) using chloroform: methanol: acetic acid (3:3:1, v/v) as a developing solvent. Products were visualized by spraying with 10 % (v/v) H2SO4 and baking at 180 °C. d-galactose, and two oligomers, neoagarotetraose and neoagarohexose prepared according to Ohta et al. (2005), were used as standards. The control experiments were done with α-agarase from Thalassomonas agarivorans JAMB-A33 (Ohta et al. 2005; Miyazaki et al. 2008) and β-agarase from Microbulbifer thermotolerans JAMB-A94 (Ohta et al. 2004; Miyazaki et al. 2008) by hydrolyzing Agarose-ME in 20 mM Tris–HCl buffer for 2, 4, 8, and 16 h at 40 °C.
Molecular mass determination
Gel-filtration chromatography was done using ÄKTAexplorer 10S System (GE Healthcare Japan) equipped with HiPrep 16/60 Sephacryl S-200 HR column (1.6 × 60 cm, GE Healthcare Japan) with a solvent (1.5 ml/min) of 3.5 M NaCl, 5 mM Tris–HCl, pH 7.0. Ribonuclease A (14.8 kDa), ovalbumin (43.5 kDa), bovine serum albumin (61.3 KDa), aldolase (176 kDa), catalase (219 kDa) were used to determine the apparent molecular weight of an agarase.
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed in a SuperSep 5–20 % polyacrylamide gradient gel (Wako Pure Chemical Industries, Ltd., Japan) with a buffer containing 25 mM Tris, 192 mM glycine, and 0.1 % SDS (pH 8.8). Proteins were stained with Bio-Safe Coomassie stain (BIO-RAD). Precision Plus dual color standards (BIO-RAD, 10–250 kDa) were run as molecular mass standards.
Results
Agarase-producing strains
A huge number of colonies appeared on agar plates from ~300 salt samples. We obtained, however, only two strains that formed depression on agar plates, and they also gave halo after flooding with Lugol’s solution (0.2 I2 plus 2 % KI). Both strains were red pigmented, coccoid forming microscopically, and did not lyse in distilled water. The 16S rRNA gene sequence (AB748563) of the higher agarolytic strain was closest to those of Halococcus salifodinae DSM 8989T (AB004877) and Halococcus saccharolyticus ATCC 49257T (AB004876) with 99.4 and 99.2 % similarity, respectively (Fig. S1). We designated the strain as Halococcus sp. 197A and the agarase from this strain as Aga-HC. Halococcus sp. 197A showed a depression with a diameter of 20 mm around a colony (about 4 mm) on agar medium after incubation at 37 °C for 2 weeks (Fig. S2). The strain was able to grow up to 45 °C, and grew optimally at around 37 °C with maximum agarase production on agar plates. The two most closely related strains Hcc. salifodinae JCM 9578T and Hcc. saccharolyticus JCM 8878T did not show any agarose-degrading activities in our experiments.
Preparation of Aga-HC
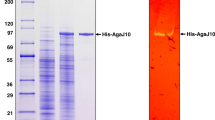
In liquid culture media, essentially no agarolytic activities were detected. Therefore, we induced the enzyme production in the HA-i medium with 0.2 % agarose as described in Materials and methods. The purified Aga-HC (~0.4 mg/ml) was homogeneous as judged from SDS-PAGE (Fig. 1). Molecular weight was estimated as 56 kDa by SDS-PAGE and 55 kDa by gel-filtration chromatography using ÄKTAexplorer 10S System. The purified Aga-HC was stable for months as long as kept in a refrigerator in 2–3 M NaCl at neutral pH.

SDS-PAGE of the purified Aga-HC in 5–20 % gradient polyacrylamide gel (lane P). Protein mass markers (in kDa) are indicated on the both side (lane M). The proteins were stained with Coomassie brilliant blue
Effects of salt and pH on agarase activity
The Aga-HC showed no activity in the presence of 0–0.3 M salt. Increasing activities were observed with the increase of NaCl concentration, with optimum at 3.5 M. In a separate experiment, KCl supported similar activities as NaCl up to 3.5 M, and LiCl to a lesser extent up to 2.5 M (Fig. 2). When these monovalent salts in the reaction mixtures were replaced by divalent cation salts, CaCl2 or MgCl2, no activity was detected at 0–0.1 M. At higher concentrations, up to 3.5 M, white precipitate [probably Ca(OH)2 or Mg(OH)2] appeared upon addition of DNS reagent, which was strongly alkaline. Absorbances (at 535 nm) of supernatants after boiling and spinning down did not increase, suggesting these divalent cations did not support activities of the agarase. The agarolytic activity was inactivated when dialysed against a NaCl-free buffer for 24 h, and recovery of activity was not observed after re-dialysis against 3.5 M NaCl, 5 mM Tris–HCl, pH 7.0 for 3 days. The enzyme showed activities from pH 4.5 to 8.5, with optimum at around pH 6 (Fig. 3).

The effect of NaCl, KCl and LiCl concentrations on the activity of Aga-HC determined at 65 °C using 0.3 % agarose in the standard buffer (50 mM MES, pH 6.0) containing 0–5.0 M NaCl and 0–3.5 M of LiCl or KCl at intervals of 0.5 M

The effect of pH on the activities of Aga-HC at 65 °C in the presence of 3.5 M NaCl. The following 50 mM buffers were used. Citric acid-NaOH (filled circle: pH 4.0–6.0), MES-NaOH (filled diamond: pH 5.5–7.0), sodium phosphate (filled square: pH 6.5–8.0), glycylglycine-NaOH (filled triangle: pH 7.0–8.5), N-tris (Hydroxymethyl) methyl-4-aminobutanesulfonic acid-NaOH (×: pH 8.5–9.5)
Effects of temperature on activity and stability
In our standard assay condition (reaction for 15 min with final agarose-ME concentration of 0.27 %), Aga-HC showed no activity at temperature lower than 35 °C, because of formation of soft gel. Increasing agarolytic activities were observed with the increase of temperature from 40 °C up to 70 °C, then decreasing activities were obtained until 100 °C (Fig. 4a). Non-enzymatic degradation of agarose was not detected. Measurement of stability of the enzyme (in 3.5 M NaCl, 5 mM Tris–HCl, pH 7.0) at high temperatures revealed its high thermostability. Aga-HC retained ~90 % of the initial activities after incubation for 1 hour at 65–80 °C (Fig. 4b). When incubated at 95 °C, more than 50 % activity remained, while almost no activity was detected after incubation at 100 °C for 30 min. In the presence of additional 10 mM CaCl2, ~17 % remaining activity was detected after 30 min at 100 °C (Fig. 4c).

Effect of temperature on the enzyme activity and stability of purified Aga-HC. The enzyme activity was measured at temperatures ranging from 35 to 100 °C using MES-NaOH buffer (pH 6.0) (a). For the thermal stability of Aga-HC, the enzyme was incubated at indicated temperatures for 0–60 min in the absence (b) or presence of 10 mM CaCl2 (c), and remaining activities were measured at 65 °C as described in “Materials and methods”
Substrate specificity and the reaction products
The enzyme showed degradation activity on agar or agarose from various suppliers as determined by the release of reducing sugar. Relative activities were expressed with that against Agarose-ME as 100 %; 97 % to Agarose-LE, 69 % to Agarose-S, 70 % to Agarose, 77 % to Bacto-agar, and 72 % to Agar noble.
Oligosaccharides in a hydrolysate of Agarose-ME at 65 °C were analyzed by TLC after desalting of the reaction mixture as described in materials and methods. Aga-HC released degradation products in the order of neoagarohexose, neoagarotetraose and small quantity of neoagarobiose (Fig. 5). The spot with slightly higher Rf value than galactose of lane 14 (after incubation for 16 h) was neoagarobiose (Ohta et al. 2005). These data indicated that Aga-HC was a β-type agarase.

Thin-layer chromatogram of hydrolysis products by three agarases on Silica 60 TLC plates (Merck). Lanes 4–8, α-agarase from Thalassomonas agarivorans JAMB-A33 (after incubation at 40 °C for 0, 2, 4, 8, 16 h), lanes 9–14, Aga-HC (after incubation at 65 °C for 0, 1, 2, 4, 8, 16 h), lanes 15–19, β-agarase from Microbulbifer thermotolerans JAMB-A94 (after 0, 2, 4, 8, 16 h at 40 °C). Markers were neoagarohexaose (lanes 1 and 22), neoagarotetraose (lanes 2 and 21), and galactose (lanes 3 and 20). A developing solvent was chloroform: methanol: acetic acid (3:3:1, v/v) and products were visualized by spraying with 10 % (v/v) H2SO4 and baking at 180 °C
Discussion
According to Stanier (1941), the first isolation of an agar-digesting bacterium was reported by HH Gran in 1902, while engaged in a general study of marine bacteria. Decomposition of agar in ocean is of undoubted importance in the cycle of matter in the ocean, where agar and similar polysaccharides form a large part of the carbohydrate constituents of many marine algae. As summarized by Fu and Kim (2010) many of agar-digesting Bacteria are of marine origin. Then, what about organisms thriving in solar saltern, a hypersaline extreme environment of seawater origin? Although numerous halophilic Archaea have been isolated from salterns and salt lakes throughout the world (Oren 2007; Enache et al. 2012), there have been no description on agar-degrading activities so far in more than 140 species belonging to Halobacteriaceae. We also have not encountered any noticeable depression of agar around colonies during our exploration for extreme halophiles possessing new phenotypic characteristics. In this study, we paid special attention to depression of more than 300 agar plates, and luckily enough we succeeded in isolating two agar-degrading strains from two solar salt samples.
At present, genome sequences of 145 Archaeal strains have been made public (Kyoto Encyclopedia of Genes and Genomes), and three strains have been suggested to possess agarase genes, Thermosphaera aggregans in the order Desulfurococcales, Thermococcus sibiricus in the order Thermococcales and Halalkalicoccus jeotgali in the order Halobacteriales. The agarase of T. aggregans and T. sibiricus have not been characterized yet (Spring et al. 2010; Mardanov et al. 2009). The haloarchaeal strain H. jeotgali JCM 14584T was reported to have been isolated from a Korean seafood produced with sea salt (Roh et al. 2007). This strain, however, showed no depression on the agar plate in our experiments. Thus, this is the first report on agarase from the domain Archaea.
The purified Aga-HC from our Archaeal strain Halococcus sp. 197A was halophilic and lost activity in the absence of NaCl as do most enzymes from halophilic Archaea (Madern et al. 2000). On the other hand, agarases from Bacteria are known to require low NaCl concentrations for their optimum activities as is the case of most other enzymes, for example 0.15 M for β-agarase II from Pseudomonas atlantica (Morrice et al. 1983) and 0.9 M for β-agarase PjaA from Pseudomonas sp. strain W7 (Ha et al. 1997). The β-agarase (Thermostable β-Agarase; Wako Pure Chemical Industries, Ltd., Japan) from Microbulbifer thermotolerans showed almost the same activities up to 1.25 M NaCl, and decreasing activities were observed with increasing NaCl concentrations. (Fig. S3a).
Most agarases of bacterial strains work optimally below 40 °C (Fu and Kim 2010). The β-agarase from Microbulbifer thermotolerans JAMB-A94 of marine origin was exceptional in that the optimum temperature was 55 °C (Ohta et al. 2004). Moderately thermophilic bacteria, Alterococcus agarolyticus, growing at 38–58 °C isolated from hot springs were reported to produce extracellular agarase on agar medium (Shieh and Jean 1998), but further studies on the strain and agarase have not been published. In our assay condition, Aga-HC exhibited no detectable activity at temperature below 35 °C since the 0.3 % agarose formed a soft gel below this temperature. Papers dealing with agarases working at low temperature have coped with this difficulty using much lower agarose concentration, e.g. 0.005 % (Morrice et al. 1983). The Aga-HC was extraordinary in showing high activities at temperature higher than 40 °C and it worked optimally at as high as 70 °C in the presence of 3.5 M NaCl. It has long been suggested that some, not all, enzymes from halophilic archaeal strains were thermophilic. In a now classical paper by Keradjopoulos and Holldorf (1977), they stated that ‘salt lakes and brines are often closed and stable hydrologic systems, in which intensive isolation and a reduced circulation cause temperatures up to 70 °C in layers of high salinity near the surface’. They demonstrated that nine enzymes of three strains of Halobacterium salinarum exhibited maxima of temperature for catalytic activities between 55 and 70 °C; those enzymes were thermophilic. Since then, a number of papers have dealt with halophilic and thermophilic enzymes from haloarachaea. For example, a serine protease from a haloarchaeal strain 172P1 (now Natrialba asiatica) exhibited high activity at 75–80 °C, when assayed in the presence of 25 % NaCl. The optimal concentration of NaCl required was 10–14 % when assayed at 70° (Kamekura and Seno 1990). A β-xylanase of a halophilic archaeon Halorhabdus utahensis exhibited optimal activity at 70 °C (Wainø and Ingvorsen 2003). More recently, recombinant alcohol dehydrogenase from the haloalkaliphilic archaeon Natronomonas pharaonis was shown to be most active at 5 M NaCl or 4 M KCl and 70 °C (Cao et al. 2008).
Aga-HC was also extraordinary in that it was quite thermostable. In the presence of 3.5 M NaCl, pH 7.0, the enzyme retained approximately 90 % of the initial activity after incubation for 1 hour at as high as 80 °C. Most agarases of bacterial strains are stable below 50 °C (Fu and Kim 2010). For example, the β-agarase from Microbulbifer thermotolerans JAMB-A94 was stable at 50 °C for 15 min in 20 mM Tris–HCl buffer (pH 7.0), while retained only 10 % activity after incubation at 70 °C for 15 min (Ohta et al. 2004). Our separate experiments showed that the enzyme was thermolabile in the presence of 2.5 M NaCl (Fig. S3b).
Likewise, β-agarases from Alteromonas sp. SY37-12 (Wang et al. 2006), Agarivorans albus YKW-34 (Fu et al. 2008), and Acinetobacter sp. Ag LSL-1 (Lakshmikanth et al. 2009) retained activities after incubation as follows; 20 % after 1 min at 70 °C, 10 % after 60 min at 70 °C, and none after 60 min at 60 °C, respectively.
The Halococcus sp. 197A that produced Aga-HC exhibiting the highest halophilicity (3.4 M NaCl for optimum), thermophilicity (70 °C for optimum) and thermostability (half-life of 60 min at 95 °C) in agarases reported so far was isolated from a solar salt sample. This fact raises some interesting and enigmatic questions; where is the indigenous habitat of the strain, what is the enzyme doing, and where did the gene of Aga-HC come from? Some haloarchaeal strains have been shown to survive in sea water (Inoue et al. 2011) and in 0.5 % NaCl solution for as long as 10 days (Fukushima et al. 2007). Halococcus species were found in the nostrils salt glands of the seabird Calonectris diomedea (Brito-Echeverría et al. 2009). Due to the restricted flow of seawater into Hamelin Pool, Shark Bay, Australia and the high net evaporation rates, the salinity of the surface water in Hamelin Pool is twice that of normal seawater and the living stromatolites are partially submerged in this hypersaline environment. Halococcus hamelinensis (Goh et al. 2006; Burns et al. 2012) was isolated from a stromatolite in Hamelin pool. Although Hcc. hamelinensis strain 106A6 is not an agarase producer, agarase-producing Halococcus strains or strains of other genera might be thriving in saline environments throughout the world. More extensive survey for agarase-producing halophiles might give some clues to the decomposition of agar in saline environments as well as in the ocean.
References
Aoki T, Araki T, Kitamikado M (1990) Purification and characterization of a novel beta-agarase from Vibrio sp. AP-2. Eur J Biochem 187:461–465
Bautista V, Esclapez J, Pérez-Pomares F, Martínez-Espinosa RM, Camacho M, Bonete MJ (2012) Cyclodextrin glycosyltransferase: a key enzyme in the assimilation of starch by the halophilic archaeon Haloferax mediterranei. Extremophiles 16:147–159
Brito-Echeverría J, López-López A, Yarza P, Antón J, Rosselló-Móra R (2009) Occurrence of Halococcus spp. in the nostrils salt glands of the seabird Calonectris diomedea. Extremophiles 13:557–565
Burmeister M, Lehrach H (1989) Isolation of large DNA fragments from agarose gels using agarase. Tren Genet 5:41
Burns BP, Gudhka RK, Neilan BA (2012) Genome sequence of the halophilic archaeon Halococcus hamelinensis. J Bacteriol 194:2100–2101
Cao Y, Liao L, Xu XW, Oren A, Wang C, Zhu XF, Wu M (2008) Characterization of alcohol dehydrogenase from the haloalkaliphilic archaeon Natronomonas pharaonis. Extremophiles 12:471–476
Cline SW, Schalkwyk LC, Doolittle WF (1989) Transformation of the archaebacterium Halobacterium volcanii with genomic DNA. J Bacteriol 171:4987–4991
Enache M, Popescu G, Itoh T, Kamekura M (2012) Halophilic microorganisms from man-made and natural hypersaline environments: physiology, ecology, and biotechnological potential. In: Stan-Lotter H, Fendrihan S (eds) Adaptation of microbial life to environmental extremes. Novel research results and application, Springer-Verlag, pp 173–197
Finkelstein M, Rownd TH (1978) A rapid method for extracting DNA from agarose gels. Plasmid 1:557–562
Fu XT, Kim SM (2010) Agarase: review of major sources, categories, purification method, enzyme characteristics and applications. Mar Drugs 8:200–218
Fu XT, Lin H, Kim SM (2008) Purification and characterization of a novel beta-agarase, AgaA34, from Agarivorans albus YKW-34. Appl Microbiol Biotechnol 78:265–273
Fukushima T, Usami R, Kamekura M (2007) A traditional Japanese-style salt field is a niche for haloarchaeal strains that can survive in 0.5% salt solution. Sal Sys 3:2
Giordano A, Andreotti G, Tramice A, Trincone A (2006) Marine glycosyl hydrolases in the hydrolysis and synthesis of oligosaccharides. Biotechnol J 1:511–530
Goh F, Leuko S, Allen MA, Bowman JP, Kamekura M, Neilan BA, Burns BP (2006) Halococcus hamelinensis sp. nov., a novel halophilic archaeon isolated from stromatolites in Shark Bay, Australia. Int J Syst Evol Microbiol 56:1323–1329
Ha JC, Kim GT, Kim SK, Oh TK, Yu JH, Kong IS (1997) β-agarase from Pseudomonas sp. W7: purification of the recombinant enzyme from Escherichia coli and the effects of salt on its activity. Biotechnol Appl Biochem 26:1–6
Hamer GK, Bhattacharjee SS, Yaphe W (1977) Analysis of the enzymic hydrolysis products of agarose by 13C-n.m.r spectroscopy. Carbohydr Res 54:C7–C10
Hu Z, Lin BK, Xu Y, Zhong MQ, Liu GM (2009) Production and purification of agarase from a marine agarolytic bacterium Agarivorans sp. HZ105. J Appl Microbiol 106:181–190
Hutcheon GW, Vasisht N, Bolhuis A (2005) Characterisation of a highly stable α-amylase from the halophilic archaeon Haloarcula hispanica. Extremophiles 9:487–495
Inoue K, Itoh T, Ohkuma M, Kogure K (2011) Halomarina oriensis gen. nov., sp. nov., a halophilic archaeon isolated from a seawater aquarium. Int J Syst Evol Microbiol 61:942–946
Kamekura M, Seno Y (1990) A halophilic extracellular protease from a halophilic archaebacterium strain 172 P1. Biochem Cell Biol 68:352–359
Keradjopoulos D, Holldorf AW (1977) Thermophilic character of enzymes from extreme halophilic bacteria. FEMS Microbiol Lett 1:179–182
Kim H-W, Ishikawa K (2010) Complete saccharification of cellulose at high temperature using endo cellulase and β-glucosidase from Pyrococcus sp. J Microbiol Biotechnol 20:889–892
Kirimura K, Masuda N, Iwasaki Y, Nakagawa H, Kobayashi R, Usami S (1999) Purification and characterization of a novel β-agarase from an alkalophilic bacterium, Alteromonas sp. E-1. J Biosci Bioeng 87:436–441
Kloareg B, Quatrano RS (1988) Structure of the cell walls of marine algae and ecophysiological functions of the matrix polysaccharides. Oceanogr Mar Biol Annu Rev 26:259–315
Lakshmikanth M, Manohar S, Lalitha J (2009) Purification and characterization of β-agarase from agar-liquefying soil bacterium, Acinetobacter sp., AG LSL-1. Proc Biochem 44:999–1003
Madern D, Ebel C, Zaccai G (2000) Halophilic adaptation of enzymes. Extremophiles 4:91–98
Mardanov AV, Ravin NV, Svetlitchnyi VA, Beletsky AV, Miroshnichenko ML, Bonch-Osmolovskaya EA, Skryabin KG (2009) Metabolic versatility and indigenous origin of the archaeon Thermococcus sibiricus, isolated from a siberian oil reservoir, as revealed by genome analysis. Appl Environ Microbiol 75:4580–4588
Miller L (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31:1321–1326
Minegishi H, Kamekura M, Kitajima-Ihara T, Nakasone K, Echigo A, Shimane Y, Usami R, Itoh T, Ihara K (2012) Gene orders in the upstream of 16S rRNA genes divide genera of the family Halobacteriaceae into two groups. Int J Syst Evol Microbiol 62:188–195
Miyazaki M, Nogi Y, Ohta Y, Hatada Y, Fujiwara Y, Ito S, Horikoshi K (2008) Microbulbifer agarilyticus sp. nov. and Microbulbifer thermotolerans sp. nov., agar degrading bacteria isolated from deep-sea sediment. Int J Sys Evol Microbiol 58:1128–1133
Morrice LM, McLean MW, Williamson FB, Long WF (1983) β-agarases I and II from Pseudomonas atlantica. Purifications and some properties. Eur J Biochem 135:553–558
Ohta Y, Nogi Y, Miyazaki M, Li Z, Hatada Y, Ito S, Horikoshi K (2004) Enzymatic properties and nucleotide and amino acid sequences of a thermostable β-agarase from the novel marine isolate, JAMB-A94. Biosci Biotechnol Biochem 68:1073–1081
Ohta Y, Hatada Y, Miyazaki M, Nogi Y, Ito S, Horikoshi K (2005) Purification and characterization of a novel α-agarase from a Thalassomonas sp. Curr Microbiol 504:212–216
Oren A (2007) Biodiversity in highly saline environments. In: Gerday C, Glansdorff N (eds) Physiology and biochemistry of extremophiles. ASM Press, Washington, pp 223–231
Pérez-Pomares F, Bautista V, Ferrer J, Pire C, Marhuenda-Egea FC, Bonete MJ (2003) Alpha-amylase activity from the halophilic archaeon Haloferax mediterranei. Extremophiles 7:299–306
Potin P, Richard C, Rochas C, Kloareg B (1993) Purification and characterization of the α-agarase from Alteromonas agarlyticus (Cataldi) comb. nov., strain GJ1B. Eur J Biochem 214:599–607
Rashid N, Cornista J, Ezaki S, Fukui T, Atomi H, Imanaka T (2002) Characterization of an archaeal cyclodextrin glucanotransferase with a novel C-terminal domain. J Bacteriol 184:777–784
Rochas C, Lahaye M, Yaphe W, Phanviet MT (1986) 13C-N.M.R.-spectroscopic investigation of agarose oligomers. Carbohydr Res 148:199–207
Roh SW, Nam YD, Chang HW, Sung Y, Kim KH, Oh HM, Bae JW (2007) Halalkalicoccus jeotgali sp. nov., a halophilic archaeon from shrimp jeotgal, a traditional Korean fermented seafood. Int J Sys Evol Microbiol 57:2296–2298
Shieh WY, Jean WD (1998) Alterococcus agarolyticus, gen. nov., sp.nov., a halophilic thermophilic bacterium capable of agar degradation. Can J Microbiol 44:637–645
Spring S, Rachel R, Lapidus A, Davenport K, Tice H, Copeland A, Cheng JF, Lucas S, Chen F, Nolan M, Bruce D, Goodwin L, Pitluck S, Ivanova N, Mavromatis K, Ovchinnikova G, Pati A, Chen A, Palaniappan K, Land M, Hauser L, Chang YJ, Jeffries CC, Brettin T, Detter JC, Tapia R, Han C, Heimerl T, Weikl F, Brambilla E, Göker M, Bristow J, Eisen JA, Markowitz V, Hugenholtz P, Kyrpides NC, Klenk HP (2010) Complete genome sequence of Thermosphaera aggregans type strain (M11TL). Stand Genom Sci 2:245–259
Stanier RY (1941) Studies on marine agar-digesting bacteria. J Bacteriol 42:527–559
Tanaka T, Fukui T, Imanaka T (2001) Different cleavage specificities of the dual catalytic domains in chitinase from the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J Biol Chem 276:35629–35635
Wainø M, Ingvorsen K (2003) Production of β-xylanase and β-xylosidase by the extremely halophilic archaeon Halorhabdus utahensis. Extremophiles 7:87–93
Wang J, Mou H, Jiang X, Guan H (2006) Characterization of a novel β-agarase from marine Alteromonas sp. SY37-12 and its degrading products. Appl Microbiol Biotechnol 71:833–839
Zhang Y, An R, Yatsunami R, Sato M, Orishimo K, Hatori Y, Fukui T, Nakamura S (2010) Characterization of a haloarchaeal GH family 18 chitinase with additional acidic amino acids on its protein surface. J Jpn Soc Extremophiles 9:72–74
Acknowledgments
Part of this study has been supported by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) by a Grant for the Strategic Development of Advanced Science and Technology S1101017 since April 2011.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by F. Robb.
Electronic supplementary material
Below is the link to the electronic supplementary material.
792_2013_575_MOESM1_ESM.tiff
Phylogenetic tree of Halococcus sp. 197A and species of the genus Halococcus based on 16S rRNA gene sequences using neighbor-joining method. The bootstrap values were generated from 1,000 replicates (TIFF 1521 kb)
792_2013_575_MOESM2_ESM.tiff
Depression of an agar plate and clearance zone after staining with Lugol’s solution (0.2 % I2 plus 2 % KI). A drop of cell suspension of Halococcus sp. 197A was placed on the center of an agar plate and incubated at 37 °C for 2 weeks (TIFF 1521 kb)
792_2013_575_MOESM3_ESM.tiff
The effect of NaCl on the enzyme activity and temperature stability of Thermostable β-Agarase from Microbulbifer thermotolerans. The enzyme activity was measured at NaCl concentrations ranging from 0.25 to 4.75 M using 5 mM Tris–HCl buffer (pH 7.0) (a) (TIFF 1521 kb)
792_2013_575_MOESM4_ESM.tiff
For the thermal stability, the enzyme was incubated at 50 °C for 0–60 min in the absence (filled circles) or presence of 2.5 M NaCl (filled square), and remaining activities were measured at 50 °C for 15 min (TIFF 1521 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Minegishi, H., Shimane, Y., Echigo, A. et al. Thermophilic and halophilic β-agarase from a halophilic archaeon Halococcus sp. 197A. Extremophiles 17, 931–939 (2013). https://doi.org/10.1007/s00792-013-0575-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-013-0575-z