
Abstract
Microarray data analysis is a major challenging field of research in recent days. Machine learning–based automated gene data classification is an essential aspect for diagnosis of gene related any malfunctions and diseases. As the size of the data is very large, it is essential to design a suitable classifier that can process huge amount of data. Deep learning is one of the advanced machine learning techniques to mitigate these types of problems. Due the presence of more number of hidden layers, it can easily handle the big amount of data. We have presented a method of classification to understand the convergence of training deep neural network (DNN). The assumptions are taken as the inputs do not degenerate and the network is over-parameterized. Also the number of hidden neurons is sufficiently large. Authors in this piece of work have used DNN for classifying the gene expressions data. The dataset used in the work contains the bone marrow expressions of 72 leukemia patients. A five-layer DNN classifier is designed for classifying acute lymphocyte (ALL) and acute myelocytic (AML) samples. The network is trained with 80% data and rest 20% data is considered for validation purpose. Proposed DNN classifier is providing a satisfactory result as compared to other classifiers. Two types of leukemia are classified with 98.2% accuracy, 96.59% sensitivity, and 97.9% specificity. The different types of computer-aided analyses of genes can be helpful to genetic and virology researchers as well in future generation.
Similar content being viewed by others

1 Introduction
The hereditary information of a human being is generally stored in the gene. Genetic disease is a type of disease that causes any disorder in the gene expressions of the human body. It happens due to the mutation in a single gene, multiple gene mutations, gene mutation combination, several environmental factors, and damages in chromosomes. Some genetic disorders or mutations can be a cause of developing cancer in the human body. Also the mutation of genes leads to a variety new generated virus. This type of change is referred to the hereditary cancer syndromes and it can be transferred from parents to a child. Leukemia is a type of bone marrow cancer that happens due to genetic disorder [1]. The lifestyle of the human being and environmental factors increase the risk of leukemia. Sometimes it may occur with no known factors. At the initial stage of leukemia, a stem cell turns into the cancerous cell and gradually it multiplies uncontrollably. These cancerous cells do not do their job well and they crowd out healthy cells in the bone marrow and in the bloodstream. The basic factor that develops leukemia in the body is the mutation of the cells in the bone marrow. Sometimes it may thwart bone marrow from developing healthy cells. It is a complicated task to detect the deoxyribonucleic acid (DNA) mutation for early diagnosis of various genetic diseases because most of the genes have many regions where mutation happens. DNA microarray is one of the advanced technologies that is used to measure the expression level of a large number of genes. This technology has the capability to determine whether the DNA of an individual having a mutation gene or not. Microarray technology is widely used for the analysis and prediction of different types of leukemia [2,3,4].
Early prediction of leukemia is a challenging job for physicians nowadays and it can be done by adopting computer-aided automated disease diagnosis systems in medical sectors. Different machine learning techniques were applied to medical datasets for developing an intelligent diagnosis system. Due to the digital revolution and advances in information technology, a huge amount of data is generating from the medical sectors. Machine learning techniques are well suited for the analysis of these large data and also several techniques were adopted for the diagnosis of different diseases [5, 6]. For successfully running any machine learning algorithms, different diagnosis data are available in the form of medical records at multiple hospitals. Due to microarray technology, a big amount of DNA expression data is also generating from hospitals. Automatic analysis and classification of these data are more essential for the early diagnosis and decision-making for any genetic disease. Research on gene expression data is one of the most popular areas in machine learning–based biomedical data analysis. Different machine learning algorithms were also used for the analysis of this data. Here in our work, we have adopted a deep learning technique for the classification of leukemia data. A total of two types of leukemia, acute lymphocyte (ALL) and acute myelocytic (AML), were classified by using deep neural networks. The classification performance of this proposed model is satisfactory as compared with earlier works as presented in the result section.
The rest of the paper is organized as follows. Related literatures are presented in Section 2. Section 3 represents the proposed work. Results obtained through the proposed model are presented in Section 4. Section 5 concludes the work.
2 Related literature
Due to the recent advancement in artificial intelligence (AI) and machine learning, different techniques were used by the researchers for the gene expression data analysis. The main goal of these machine learning techniques is to increase the performance of the model by increasing the accuracy and decreasing the error. Authors in [7] adopted three different tree-based supervised machine learning algorithms for classifying cancer from gene expression data. A total of seven types of cancer data were classified using the C4.5 decision tree and bagged and boosted decision trees. From their result, it can be observed that the bagging decision tree classifier is performing better as compared with the other two. The selection of suitable features from the original data is a challenging task in the machine learning approach and it is more important in biomedical data. In order to improve the performance, the authors have used the null space-based feature selection method in [8]. The redundant gene expressions were discarded by null space information generated through scatter matrices. After successfully reducing the feature dimension, the classification was done by applying three types of classifiers such as support vector machine (SVM), Naive Bayes, and linear discriminant analysis (LDA). For identifying AML samples, a one-class SVM classifier was applied in gene expression data in [9]. Their proposed classifier’s performance was verified with different types of kernel function and it was observed that the accuracy was better with a linear kernel. They have also compared their obtained result with some earlier traditional classification models and they have claimed that their result was better as compared with others. Chi-square feature selection technique was introduced to deal with the high-dimension problem in [10]. In their work, they have applied machine learning techniques in the SAGE data set. From the result, they have claimed that SVM and Naive Bayes classifiers were providing better results with the reduced features. The performance of the extreme learning machine (ELM) algorithm was also evaluated with the gene expression dataset in [11]. The reasons for adopting the ELM algorithm were to improve the training time, avoiding local minima and overfitting problems. The multicategory classification was performed in three different types of microarray datasets. Authors in [12] used the random forest algorithm for the selection of gene patterns. The aim of their proposed model was to find significant genes with increased accuracy. Authors have also considered three different types of gene expression datasets for validating the performance of the machine learning model. Their proposed hybrid model was providing efficient result as compared with the traditional models. Another hybrid model for selecting gene patterns was proposed in [13]. They have applied hierarchical clustering and rough set theory approach for the classification of gene patterns. Their proposed approach was a three-step structure. Initially, the gene clusters were formed by using a hierarchical method. After this, the initial clusters were again divided into more clusters by applying lower and upper approximation property of the rough set algorithm. Gene ranking and cluster ranking method were applied to rank the clusters for selecting significant genes. After completing these preprocessing steps, the classification was done by using SVM classifier. In order to design an optimized gene data classification model, authors in [14] have used multi-objective spotted hyena optimizer (MOSHO) and salp swarm algorithm (SSA) in their work. As compared with the real-life optimizing algorithm, the proposed SSA technique maintains the diversity and training information. MOSHA technique needs low computational time for maintaining the required information. For classification purposes, they have used four types of classifiers in their work. Again SVM classifier was used for classifying the gene expressions in [15,16,17,18]. Authors in [17] applied the gray wolf optimization algorithm to develop an optimized data mining model for gene expression data classification. In their work, they have applied information gain techniques for feature selection. Breast and colon cancer gene data was used for the classification purpose. Correlation-based gene features were selected for classification in [18]. For selecting the optimized parameters of the SVM classifier, they have applied teaching-learning-based optimization (TLBO) algorithm in their proposed work. For converting the continuous version of the i = optimized parameter to binary, they have selected a new encoding technique. K-nearest neighbor (KNN) is another popular data mining method which was broadly used in gene expression data classification [18,19,20,21,22]. The combination of KNN and SVM classifier was proposed in [18, 19] for classifying the leukemia and colon cancer data. Authors in [20] used particle swarm optimization (PSO) and KNN for classifying AML and ALL gene expressions. Different gene subsets were obtained by applying genetic algorithm in [21] and KNN was used for classifying the subsets.
The neural network and its variants were also deployed for the analysis of gene expression in various studies. An artificial neural network (ANN) was used for classifying the gene expression in [23,24,25,26]. Authors in [25] preprocessed the gene data by applying normalization and feature selection techniques. K-means clustering technique was applied for creating the specific gene clusters. For reducing the feature size, they have applied SVM in their work. Finally, the reduced selective features were classified using ANN. The proposed model was validated with two types of tumor dataset. A supervised machine learning approach was proposed for classifying the ALL and AML gene data was proposed in [26]. The classification result was better in the ANN classifier as compared with other classifiers as they have claimed. A radial basis function network (RBFN) was used to classify the gene sequences. A novel rough set-based feature selection technique was presented in [27]. The benefit of adopting this method was to avoid gene expression clustering. They have combined the roughest feature selection technique and RBFN for predicting leukemia, lung cancer, and prostate cancer. In their work, they have validated their result with the other two types of classifiers and from the result, it can be observed that RBFN was providing better results as compared with LSVM and Naive Bayes. A generalized RBFN was used for improving the prediction accuracy in gene classification. The efficient features were selected by using BARS, BIRS, and FCBF techniques. Functional logistic discriminant analysis (FLDA) and RBFN were used for the analysis of microarray gene expression data in [28]. Probabilistic neural network (PNN) is another type of neural network which has been used for the analysis and classification of gene expression [29]. After selecting the effective gene features, the classification was conducted by using PNN. The performance of PNN was compared with KNN for classifying three types of data.
Deep learning is an advanced neural network technique that has been widely used in different biomedical applications. Also, researchers have used different deep learning approaches in gene expression data analysis. The relevant gene features were extracted by applying a convolutional neural network in [30]. The extracted convolutional features were then classified by using SVM. ALL leukemia was classified by applying deep learning–based image processing techniques in [31]. After collecting the microarray image, the segmentation step was done for improving the performance. After completing the segmentation steep, a seven-layer convolutional network was used for the classification purpose. A hybrid approach by combining CNN and SVM was introduced by the authors to classify the bone marrow expression images in [32]. A deep convolutional neural network was used for classifying leukemia in [33]. For successfully training the model, they have used a pertained AlexNet model for their work. A total of four types of leukemia were classified by using CNN. In another study, authors have used a hybrid deep learning model for classifying the leukemia cells. Their proposed hybrid approach was capable of extracting efficient features from the input image. They have used the global average pooling approach in their work to improve the performance. The blood cell images were classified by applying the CNN in [34]. Authors have also used the same seven-layer CNN structure for the classification of leukemia. A discrete cosine transform (DCT)–based CNN classifier model was developed by the authors in [35]. For extracting the features, they have applied DCT in the microscopic images and then classification was conducted by using CNN.
From the literature, it can be observed that a numerous amount of work were conducted for gene expression data classification. Different machine learning algorithms were used to getting a satisfactory accuracy. For reducing the computational time and increasing the accuracy, researchers have also used a deep learning–based approach in gene expression data. Most of the deep learning–based works were conducted with microarray images. But here in this work, we have taken an attempt to classify the previously extracted records of gene features and also we have achieved a better result with this data as compared with other classification models.
3 Proposed method
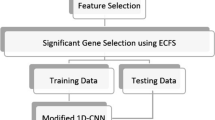
The proposed deep learning–based leukemia classification model consists of three steps as presented in Fig. 1. After collecting the data, it is preprocessed. In the preprocessing step, the entire dataset is checked for detecting the missing value because it may hamper the classification result. Here in our dataset, there was no missing value. After checking the missing value, the entire dataset is divided into two subsets. A deep neural network (DNN) classifier is used for classification purpose.

The architecture of the proposed model
3.1 Dataset and preprocessing
The leukemia dataset is collected from the publicly available microarray gene data [36]. The dataset contains the bone marrow expressions of 72 samples with 7128 genes. There are two types of leukemia classes that are there in the dataset. Forty-seven samples are belonging to ALL class and rest 25 are of AML class with zero missing values. After checking for the missing values, the entire data set is divided into training and testing purposes. Around 80% (58 samples) of data is separated for training purpose and rest 20% (18 samples) data is used for testing and validation.
3.2 Classification model
The deep neural network is an advanced neural network model composed of multiple layers where input data is represented with different levels of abstraction. Due to the hierarchical structure of the DNN, it is very easier to represent complex problems. The multiple hidden layers presented in the DNN can be the capability to learn the features from the input data. After learning the features through the hidden layers, the classification is done in the last layer of DNN. The structure of the simple DNN is presented in Fig. 2. In our proposed DNN classifier, there are of five-layer with three hidden layers. DNN is a type of feed-forward neural network where the input data flows from the input layer to the output layer without forwarding back. In the first stage, a map of a virtual neuron is created with random values or weights to make the connections between them. Then, the weights are multiplied with the input data and return the output between 0 and 1. The weights are adjusted with each other if the network faces problem at the time of recognizing a particular pattern [37].

Proposed DNN structure
By employing pooling operations and a set of distinct moving filters, CNNs seize correlations between adjacent data pieces, and then generate a successively higher level abstraction of the input data. CNNs can extract features while reducing the model complexity, which mitigates the risk of overfitting. These characteristics make CNNs achieve remarkable performance in signal processing. The vanishing gradient problem can be partially ameliorated by using non-saturating activation functions such as rectified linear unit (ReLU). Each block coordination–based learning algorithm can guarantee to globally converge.
In the proposed DNN, the input gene data G = {g1, g2, g3, ……, gn} is multiplied with the weight W = {w1, w2, w3, ………, wn}. The output (O) of each layer after multiplying with the weight is represented as
where b is the bias. After completing the weight multiplication with input data, it is passed through the nonlinear activation function and can be represented by:
where Y is the output of DNN and n is each input gene feature. f is the activation function used in the hidden layer and output layer. Here in our DNN, we have used the rectified linear unit (ReLU) activation function in the hidden layer and softmax in the output layer. ReLU is one of the most used activation function in the deep neural network. The main advantage of using this activation function is that it does not activate all the neurons at the same time and converts all the negative input into zero so that the neuron does not get activated. Training performance in this activation function is much faster as compared with other activation function [38]. It can be represented as
where g is the input data and f(g) is the output function that returns the maximum value between 0 and input data. In softmax activation function, the exponential (e-power) of the given input value and the sum of exponential values of all the values in the inputs are computed. Then, the ratio of the exponential of the input value and the sum of exponential values is the output of the softmax function. The main advantage of using it is the output probabilities range. The range varies between 0 and 1 and the sum of all the probabilities will be equal to one. If the softmax function is used for the multi-classification model, it returns the probabilities of each class and the target class will have a high probability. Mathematically softmax activation function can be represented as:
where the input is x and the output value of s is between 0 and 1 and their sum is equal to 1.
4 Results
The microarray data set is generally represented with a x by y matrix where x represents the total number of samples and y represents the gene features. Among 72 samples, around 80% of data (56 samples) are separated for training the DNN and the rest 20% (16 samples) are stored for validation purposes. The detailed structure of the training and testing sets is presented in Table 1.
The deep neural network is trained with the training data. After successfully completing the training, the proposed classifier is validated with the testing data. The training performance is presented in Table 2 and testing performance is presented in Table 3. Figures 3 and 4 show the accuracy and the loss of the proposed DNN classifier with testing data. Figure 5 shows the final graphical representation of the DNN performance.

Testing accuracy of the DNN classifier

Testing loss of the proposed DNN classifier

DNN performance for gene expression data classification
5 Discussions
For the comparison of the proposed result, we have compared our result with the other three types of classifiers. The results are presented in Table 4 and Fig. 6. The performance of the DNN classifier is compared with SVM, KNN, and Naive Bayes classifiers. From the result, it can be observed that our proposed DNN classifier is performing better as compared with the other three. In Table 5, the obtained result is compared with some earlier work and it can be observed that our proposed deep learning classifier is performing better for classifying gene expression from the microarray data.

Performance comparison of proposed DNN with SVM, KNN, and Naive Bayes classifiers
6 Conclusion
Nowadays, genetic disease is one of the major health problems worldwide. Lots of people are suffering from this disease. Early detection and diagnosis of this illness can save life. The proposed work tries to classify the leukemia types by using the deep learning approach. As the size of the gene expression data is very large, a seven-layer deep neural network is designed to classify two types of leukemia. The classification accuracy is quite better as compared with other types of classifiers. The advantage of using deep learning–based classifier is to improve the classification accuracy with minimum computational time. Around 98% classification accuracy is found from the DNN classifier and it is better than SVM, KNN, and Naive Bayes classifiers. This approach can be a useful machine learning tool for automated analysis of microarray data analysis. In the future, the accuracy can be improved by optimizing the network structure. Also the study of mutation of gene recognition will support the genetic as well as virology researchers. Like the current scene of COVID-19 would be early recognized and diagnosed.
References
Bracher-Smith M, Crawford K, Escott-Price V (2020) Machine learning for genetic prediction of psychiatric disorders: a systematic review. Mol Psychiatry:1–10
Taub FE, DeLeo JM, Thompson EB (1983) Sequential comparative hybridizations analyzed by computerized image processing can identify and quantitate regulated RNAs. Dna 2(4):309–327
Goswami RS, Sukhai MA, Thomas M, Reis PP, Kamel-Reid S (2009) Applications of microarray technology to acute myelogenous leukemia. Cancer informatics 7:CIN. S1015
Ratley A, Minj J, Patre P (2020) Leukemia disease detection and classification using machine learning approaches: a review. In: 2020 First International Conference on Power, Control and Computing Technologies (ICPC2T). IEEE, pp 161–165
Kononenko I (2001) Machine learning for medical diagnosis: history, state of the art and perspective. Artif Intell Med 23(1):89–109
Mahmood N, Shahid S, Bakhshi T, Riaz S, Ghufran H, Yaqoob M (2020) Identification of significant risks in pediatric acute lymphoblastic leukemia (ALL) through machine learning (ML) approach. Medical & Biological Engineering & Computing:1–10
Tan AC, Gilbert D (2003) Ensemble machine learning on gene expression data for cancer classification
Sharma A, Imoto S, Miyano S, Sharma V (2012) Null space based feature selection method for gene expression data. Int J Mach Learn Cybern 3(4):269–276
Vasighizaker A, Sharma A, Dehzangi A (2019) A novel one-class classification approach to accurately predict disease-gene association in acute myeloid leukemia cancer. PLoS One 14(12):e0226115
Jin X, Xu A, Bie R, Guo P (2006) Machine learning techniques and chi-square feature selection for cancer classification using SAGE gene expression profiles. In: International Workshop on Data Mining for Biomedical Applications. Springer, pp 106–115
Zhang R, Huang G-B, Sundararajan N, Saratchandran P (2007) Multicategory classification using an extreme learning machine for microarray gene expression cancer diagnosis. IEEE/ACM transactions on Computational Biology and Bioinformatics 4(3):485–495
Mallick PK, Mishra D, Patnaik S, Shaw K (2016) A semi-supervised rough set and random forest approach for pattern classification of gene expression data. International Journal of Reasoning-based Intelligent Systems 8(3–4):155–167
Mallick PK, Mishra D, Patnaik S, Shaw K (2016) A hybrid approach for simultaneous gene clustering and gene selection for pattern classification. Indian J Sci Technol 9(21):1–10
Sharma A, Rani R (2019) C-HMOSHSSA: gene selection for cancer classification using multi-objective meta-heuristic and machine learning methods. Comput Methods Prog Biomed 178:219–235
Alanni R, Hou J, Azzawi H, Xiang Y (2019) A novel gene selection algorithm for cancer classification using microarray datasets. BMC Med Genet 12(1):10
Maniruzzaman M, Rahman MJ, Ahammed B, Abedin MM, Suri HS, Biswas M, El-Baz A, Bangeas P, Tsoulfas G, Suri JS (2019) Statistical characterization and classification of colon microarray gene expression data using multiple machine learning paradigms. Comput Methods Prog Biomed 176:173–193
Loey M, Jasim MW, El-Bakry HM, Taha MHN, Khalifa NEM (2020) Breast and colon cancer classification from gene expression profiles using data mining techniques
Shukla AK, Singh P, Vardhan M (2019) A new hybrid wrapper TLBO and SA with SVM approach for gene expression data. Inf Sci 503:238–254
Mei Z, Shen Q, Ye B (2009) Hybridized KNN and SVM for gene expression data classification. Life Science Journal 6(1):61–66
Kar S, Sharma KD, Maitra M (2015) Gene selection from microarray gene expression data for classification of cancer subgroups employing PSO and adaptive K-nearest neighborhood technique. Expert Syst Appl 42(1):612–627
Li L, Darden TA, Weingberg C, Levine A, Pedersen LG (2001) Gene assessment and sample classification for gene expression data using a genetic algorithm/k-nearest neighbor method. Comb Chem High Throughput Screen 4(8):727–739
Kumar M, Rath SK (2014) Microarray data classification using fuzzy K-nearest neighbor. In: 2014 International Conference on Contemporary Computing and Informatics (IC3I). IEEE, pp 1032–1038
Chen W, Lu H, Wang M, Fang C (2009) Gene expression data classification using artificial neural network ensembles based on samples filtering. In: 2009 International Conference on Artificial Intelligence and Computational Intelligence. IEEE, pp 626–628
Pradhan M (2018) Prediction using ANN-based classifier in DNA microarray. Int J Appl Res Inf Technol Comput 9(1):1–14
Mabu AM, Prasad R, Yadav R (2020) Gene expression dataset classification using artificial neural network and clustering-based feature selection. International Journal of Swarm Intelligence Research (IJSIR) 11(1):65–86
Dwivedi AK (2018) Artificial neural network model for effective cancer classification using microarray gene expression data. Neural Comput & Applic 29(12):1545–1554
Chiang J-H, Ho S-H (2008) A combination of rough-based feature selection and RBF neural network for classification using gene expression data. IEEE transactions on nanobioscience 7(1):91–99
Araki Y, Konishi S, Imoto S (2004) Functional discriminant analysis for microarray gene expression data via radial basis function networks. In: Proceedings of COMPSTAT Symposium. pp. 613–620
Huang C-J (2004) Class prediction of cancer using probabilistic neural networks and relative correlation metric. Appl Artif Intell 18(2):117–128
Huynh P-H, Nguyen V-H, Do T-N (2019) Novel hybrid DCNN–SVM model for classifying RNA-sequencing gene expression data. Journal of Information and Telecommunication 3(4):533–547
Rehman A, Abbas N, Saba T, Rahman SI, Mehmood Z, Kolivand H (2018) Classification of acute lymphoblastic leukemia using deep learning. Microsc Res Tech 81(11):1310–1317
Vogado LH, Veras RM, Araujo FH, Silva RR, Aires KR (2018) Leukemia diagnosis in blood slides using transfer learning in CNNs and SVM for classification. Eng Appl Artif Intell 72:415–422
Shafique S, Tehsin S (2018) Acute lymphoblastic leukemia detection and classification of its subtypes using pretrained deep convolutional neural networks. Technology in cancer research & treatment 17:1533033818802789
Thanh T, Vununu C, Atoev S, Lee S-H, Kwon K-R (2018) Leukemia blood cell image classification using convolutional neural network. International Journal of Computer Theory and Engineering 10(2):54–58
Kant S, Kumar P, Gupta A, Gupta R (2018) LeukoNet: DCT-based CNN architecture for the classification of normal versus leukemic blasts in B-ALL Cancer. arXiv preprint arXiv:181007961
Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA (1999) Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 286(5439):531–537
Schmidhuber J (2015) Deep learning in neural networks: an overview. Neural Netw 61:85–117
Haykin S (1994) Neural networks: a comprehensive foundation. Prentice Hall PTR
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mallick, P.K., Mohapatra, S.K., Chae, GS. et al. Convergent learning–based model for leukemia classification from gene expression. Pers Ubiquit Comput 27, 1103–1110 (2023). https://doi.org/10.1007/s00779-020-01467-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00779-020-01467-3