
Abstract
Alanyl-peptide nucleic acid (alanyl-PNA)/DNA chimeras are oligomers envisaged to be beneficial in efficient DNA diagnostics based on an improved molecular beacon concept. A synthesis of alanyl-PNA/DNA chimera can be based on the solid phase assembly of the oligomer with mixed oligonucleotide/peptide backbone under DNA synthesis conditions, in which the nucleotides are introduced as phosphoramidites, whereas the nucleo amino acids make use of the acid labile monomethoxytrityl (MMT) group for temporary protection of the α-amino groups and acyl protecting groups for the exocyclic amino functions of the nucleobases. In this work, we realized for the first time the synthesis of all four MMT/acyl-protected nucleo alanines, achieved by deprotection/reprotection of the newly synthesized Boc/acyl intermediates, useful monomers for the obtainment of (alanyl-PNA)/DNA chimeras by conditions fully compatible with the standard phosphoramidite DNA synthesis strategy.
Similar content being viewed by others

Introduction
The unambiguous and fast detection of oligonucleotide sequences in vitro and in vivo is currently a major goal especially since more and more diseases are linked to their genetic origin. In 1996 Tyagi and Kramer introduced the concept of a molecular beacon (Fig. 1a) (Tyagi and Kramer 1996; Tyagi et al. 1998) as one of many diagnostic methods for DNA single strand sequence detection, as well as mismatch identification, known so far (Pfeifer 1996; Taylor 1997; Didenko 2002). The molecular beacon is a DNA single strand composed of a loop region having a length of around 20 nucleotides with a sequence complementary to the target DNA and a stem region in which about ten terminal nucleotides are paired in a double strand (Fig. 1a) (Tan et al. 2000). On the 5′- or 3′-terminus, respectively, a fluorophore and a quencher are linked. Fluorescence is quenched as long as the stem region is paired in a double strand. Recognition of the target DNA by formation of a rigid helical double strand leads to separation of the termini which can easily be detected by fluorescence. Besides the genetic analysis (Kostrikis et al. 1998; Socher et al. 2008) and detection of point mutations (Kranaster et al. 2008; Ficht et al. 2004; Valis et al. 2005), the molecular beacon concept is also useful in real-time PCR (Tyagi and Kramer 1996; Tyagi et al. 1998), RNA detection in living cells (Matsuo 1998), and also in the identification of protein–DNA interactions (Li et al. 2000). The introduction of single molecule detection for the interaction of a molecular beacon with DNA leads to an increase in sensitivity from about 10−7 M to 10−11 to 10−12 M (Knemeyer and Sauer 2000). Nevertheless, the sensitivity based on this detection is limited due to the solvent accessibility of the fluorophore.

In order to gain even higher sensitivity for DNA detection, we propose to use an alanyl-peptide nucleic acid (alanyl-PNA) double strand as the stem of a molecular beacon keeping the DNA recognition loop (Fig. 1a). Alanyl-PNA is an artificial oligomer that forms specific and stable double strands based on nucleobase recognition (Diederichsen 1996). The alanyl-PNA pairing system is orthogonal to oligonucleotides with respect to double strand formation and provides a possibility to hide the fluorophore from polar solvent (Diederichsen and Weicherding 1999; Küsel et al. 2005).
An alanyl-PNA oligomer is based on a regular peptide backbone composed of alanyl amino acids that carry covalently linked nucleobases in β-position of the side chain (Fig. 2) (Diederichsen 1997a, b). Oligomerization of the nucleo amino acids allows for the formation of rigid and well defined double strands with linear topology based on base pair recognition, stacking, and solvation. Base pairs are stacking with a distance of about 3.5 Å providing a completely extended peptide backbone. Nevertheless, intercalation can be made possible by creating oligomers with a missing base pair by introduction of glycines in opposite positions (Diederichsen 1997b). Evidence for a non-polar environment within the base stack of alanyl-PNA is derived from a stabilizing effect by incorporation of aromatic amino acids in alanyl-PNA (Diederichsen and Weicherding 1999). Shielding of the fluorophore from the water environment when intercalated within the alanyl-PNA duplex should be a major advantage of the chimeric alanyl-PNA/DNA molecular beacon. To ensure for a stoichiometrically well defined system, the fluorophore will be covalently linked to the peptide backbone. Guanine nucleobases in the complementary alanyl-PNA strand will function as quencher (Knemeyer and Sauer 2000).

Linear alanyl-PNA double strand: a fluorophore can be hidden within the nucleobase stack covalently linked to an alanyl side chain instead of a nucleobase
Materials and methods
General Remarks
The solvents used in this work were of the highest grade available. All reagents were of analytical grade and used without further purification. Optical rotations were determined with a Perkin-Elmer 241 polarimeter. IR spectra were recorded with a Perkin-Elmer 1600 Series FT-IR spectrometer using KBr pellets. NMR spectra were recorded with a Varian INOVA-500 instrument or a Varian Unity 300 instrument. Chemical shifts are referenced to the residual solvent peaks of [D6]DMSO (1H: δ = 2.49 ppm; 13C: δ = 39.5 ppm). ESI-MS data were measured with a LCQ Finnigan spectrometer. HRMS data were determined with a Bruker APEX-Q IV 7T spectrometer. Chromatographic separations were performed using silica gel 60 (0.040–0.063 mm, Merck). HPLC analysis was performed using either a Beckman System Gold or Pharmacia Äkta Basic using YMC-Pack ODS, RP-C18 columns for analytical samples (250 mm × 4.6 mm, 5 μm, 120 Å) with a linear gradient of A (0.1% TFA in H2O) to B (MeCN/H2O, 9:1 + 0.1% TFA). The optical purity was determined by HPLC analysis of the amino acid dimers obtained with N-Boc-l-Ala-OSu. Analytical thin-layer chromatography was performed by using Merck silica gel 60 F254 precoated aluminum plates and visualized with UV light (254 nm) or by dyeing with ninhydrin (3% in ethanol).
(l)-N α -tert-Butoxycarbonyl-β-[N6-(anisoyl)adenin-9-yl]alanine (7)
DBU (1.17 g, 7.66 mmol) was added to a solution of N6-anisoyl-adenine 8 (3.10 g, 11.5 mmol) in DMSO (40 ml). A solution of Boc-l-serine lactone 5 (1.43 g, 7.66 mmol) in DMSO (6 ml) was slowly added and stirred for 72 h at room temperature. The reaction was terminated by addition of AcOH (1.00 ml), the solvent removed, and the resulting oil coevaporated with toluene. The residue was dissolved in EtOAc and purified by flash chromatography (4 cm × 25 cm, EtOAc/MeOH/H2O/AcOH = 160:28:12:1). Removal of the solvent, repeated coevaporation with toluene, and freeze drying provided product 7 as white solid (1.13 g, 32%). The identity of monomer 7 was confirmed by comparison of the obtained 13C NMR data to those published by Howarth and Wakelin (1997) for adenine-containing N7 and N9-regioisomers. TLC R f : 0.71 (CHCl3:MeOH:H2O:HOAc = 7:3:0.3:0.03); ee 92% (HPLC: t R 30.5 min, 5–50% B in 45 min); \( \left[ \alpha \right]_{\text{D}}^{20} \) = +16.3° (c 0.08, MeOH); IR (KBr): \( \tilde{\nu } \) = 3,426, 2,926, 1,697, 1,608, 1,458 cm−1; 1H NMR (300 MHz, [D6]DMSO, 35°C) δ = 10.90 (s, br, 1H, CO2H), 8.63 (s, 1H, H-2), 8.26 (s, 1H, H-8), 8.02 (d, 3 J H,H = 8.7 Hz, 2H, Ar), 7.04 (d, 3 J H,H = 8.7 Hz, 2H, Ar), 6.09 (m, 1H, Boc-NH), 4.77 (m, 1H, H-β), 4.47 (m, 1H, H-α), 4.08 (m, 1H, H-β), 3.84 (s, 3H, OCH3), 1.05–1.35 (m, 9H, Boc); 13C NMR (75 MHz, [D6]DMSO, 35°C) δ = 171.3, 164.9, 162.4, 154.6, 152.8, 150.9, 149.9, 144.8, 130.4, 125.8, 124.8, 113.6, 77.6, 55.4, 45.5, 28.0; MS (ESI) m/z 455.8 [M − H]−; HRMS (ESI): calcd. for C21H25N6O6: 457.1830; found 457.1835 [M + H]+.
(l)-N α -tert-Butoxycarbonyl-β-[N4-(4-tert-butylbenzoyl)cytosin-1-yl]alanine (10)
DBU (1.83 g, 12.0 mmol) was added at room temperature to a solution of (4-tert-butylbenzoyl)cytosine 9 (2.28 g, 8.41 mmol) in DMSO (72 ml). A solution of Boc-l-serine lactone 5 (1.50 g, 8.01 mmol) in DMSO (18 ml) was added dropwise. After stirring overnight at room temperature the reaction was terminated by addition of AcOH (1.00 ml). The solvent was removed. The resulting oil was coevaporated with toluene before purification by repeated flash chromatography (4 cm × 22 cm, AcOEt/MeOH/H2O/AcOH = 160:28:12:1). Removal of solvent was followed by repeated coevaporation with toluene, and freeze drying led to amino acid 10 (1.53 g, 42%). TLC R f 0.60 (CHCl3:MeOH:H2O:AcOH = 7:3:0.3:0.03); ee 99% (HPLC: t R 30.6 min, 5–75% B in 40 min); \( \left[ \alpha \right]_{\text{D}}^{20} \) = −106.3° (c 0.09, MeOH); IR (KBr): \( \tilde{\nu } \) = 3,410, 2,925, 1,682, 1,607, 1,455 cm−1; 1H NMR (300 MHz, [D6]DMSO, 35°C) δ = 10.95 (s, br, 1H, CO2H), 7.93 (m, 3H, Ar, H-6), 7.50 (d, 3 J H,H = 8.7 Hz, 2H, Ar), 7.18 (d, 3 J H,H = 7.1 Hz, 1H, H-5), 6.37 (m, 1H, Boc-NH), 4.58 (m, 1H, H-β), 4.16 (m, 1H, H-α), 3.55 (m, 1H, H-β), 1.20–1.40 (m, 18H, tBu, Boc); 13C NMR (75 MHz, [D6]DMSO, 35°C) δ = 171.8, 167.0, 162.8, 155.6, 155.0, 150.4, 146.8, 130.5, 128.2, 125.1, 95.3, 78.0, 61.5, 51.8, 34.7, 30.8, 28.0; MS (ESI) m/z 481.5 [M + Na]+; HRMS (ESI): calcd. for C23H31N4O6: 459.2238; found 459.2241 [M + H]+.
(l)-N α -tert-Butoxycarbonyl-β-[(N2-isobutyryl)-guanin-9-yl]-alanine (11)
DBU (2.23 g, 14.7 mmol) was added at room temperature to a solution of (isobutyryl)guanine 12 (4.12 g, 17.4 mmol) in DMSO (15 ml). A solution of Boc-l-serine lactone 5 (2.50 g, 13.4 mmol) in DMSO (10 ml) was slowly added and stirred overnight at room temperature. The solvent was removed and the residue purified by chromatography (5 cm × 38 cm, DCM/MeOH/EtOH = 6:3:1). The fractions were collected, concentrated and investigated by NMR. The fractions containing the desired product, identified by comparison of the obtained 13C NMR data with those previously published for N9-regioisomer (Kovács and Schmél 2000; Rosenbohm et al. 2004), were combined and the solvent evaporated to dryness. After removal of the solvent product 11 (1.86 g, 31%) was obtained as a white solid. TLC R f 0.66 (CHCl3:MeOH:H2O:AcOH = 7:3:0.3:0.03); ee 96% (HPLC: t R 27.0 min, 10–50% B in 50 min); \( \left[ \alpha \right]_{\text{D}}^{20} \) = −60.0° (c 0.1, MeOH); IR (KBr): \( \tilde{\nu } \) = 3,425, 2,976, 1,682, 1,609, 1,565, 1,409 cm−1; 1H NMR (500 MHz, [D6]DMSO, 35°C) δ = 12.05 (s, br, 1H, NH), 11.68 (s, br, 1H, CO2H), 7.78 (s, 1H, H-8), 7.16 (m, 1H, Boc-NH), 4.47 (m, 1H, H-β), 4.34 (m, 1H, H-α), 4.22 (m, 1H, H-β), 2.79 (sept., 3 J H,H = 6.8 Hz, 1H, iBu), 1.15–1.36 (m, 9H, Boc), 1.11 (d, 3 J H,H = 6.8 Hz, 6H, iBu); 13C NMR (125 MHz, [D6]DMSO, 35°C) δ = 180.1, 171.1, 155.2, 154.8, 148.7, 147.8, 140.0, 119.9, 78.4, 53.1, 43.7, 34.6, 28.0, 18.8; MS (ESI) m/z 431.4 [M + Na]+; HRMS (ESI): calcd. for C17H25N6O6: 409.1830; found 409.1829 [M + H]+.
(l)-N α -(4-Monomethoxytrityl)-β-[thymin-1-yl]alanine (1)
In TFA (2 ml) Boc-β-[thymine-1-yl]alanine 6 (103 mg, 329 μmol) was dissolved and stirred at room temperature for 2 h. TFA was removed in vacuo and repeatedly coevaporated with toluene. After lyophilization from H2O the residue was dissolved in DMF (40 ml). Triethylamine (133 mg, 1.32 mmol) and 4-monomethoxytritylchloride (102 mg, 329 μmol) in DMF (2 ml) were added dropwise. After stirring at room temperature for 1 h the solvent was removed and the remaining oil was extracted three times with DCM/H2O (2:1, 200 ml). The organic phase was dried over Na2SO4 and concentrated in vacuo. Diethyl ether (3 ml) was added and the suspension stirred at −28°C for 1 h. After centrifugation at −10°C and separation from diethyl ether a white solid was obtained. This procedure was repeated to give the amino acid 1 (65.6 mg; 41%). TLC R f 0.70 (DCM:MeOH:NEt3 = 4:1:0.05); \( \left[ \alpha \right]_{\text{D}}^{20} \) = +35.5° (c 0.07, CHCl3); IR (KBr): \( \tilde{\nu } \) = 3,440, 1,697, 1,668, 1,509 cm−1; 1H NMR (500 MHz, [D6]DMSO, 35°C) δ = 11.30 (s, br, 1H, CO2H), 7.73 (s, 1H, H-6), 7.13–7.30 (m, 12H, MMT), 6.77 (d, 3 J H,H = 9.0 Hz, 2H, MMT), 5.73 (s, 1H, NH), 3.87 (s, 1H, H-β), 3.71 (s, 3H, MMT–OCH3), 3.57 (m, 1H, H-β), 3.42 (m, 1H, H-α), 1.83 (s, 3H, CH3); 13C NMR (125 MHz, [D6]DMSO, 35°C) δ = 173.6, 164.3, 157.5, 150.9, 141.0, 146.2, 143.0, 137.4, 129.7, 128.1, 126.2, 113.0, 107.5, 69.6, 55.2, 54.9, 50.7, 11.8; MS (ESI) m/z 484.6 [M − H]−; HRMS (ESI): calcd. for C28H28N3O5: 486.2024; found 486.2028 [M + H]+.
(l)-N α -(4-Monomethoxytrityl)-β-[N6-(anisoyl)adenin-9-yl]alanine (2)
In TFA (2 ml) Boc-β-[N6-(anisoyl)adenine-9-yl]alanine 7 (100 mg, 219 μmol) was dissolved and stirred at room temperature for 1 h. TFA was removed in vacuo and repeatedly evaporated with toluene. After lyophilization from H2O the residue was dissolved in DMF (25 ml). Triethylamine (89.1 mg, 876 μmol) and 4-monomethoxtritylchloride (136 mg, 440 μmol) in DMF (4 ml) were added, stirred overnight at room temperature, and concentrated in vacuo. The obtained oil was extracted Three times with DCM/H2O (2:1, 200 ml), the organic phase was dried over Na2SO4, and the solvent removed. Diethyl ether (3 ml) was added and the suspension stirred at −28°C for 1 h. Centrifugation at −10°C and separation from the diethyl ether phase yielded amino acid 2 (92.0 mg, 67%) after repeated extraction. TLC R f 0.27 (AcOEt:MeOH = 7:3); \( \left[ \alpha \right]_{\text{D}}^{20} \) = +28.3° (c 0.1, CHCl3); IR (KBr): \( \tilde{\nu } \) = 3,430, 2,928, 1,704, 1,608, 1,458 cm−1; 1H NMR (500 MHz, [D6]DMSO, 35°C) δ = 11.99 (s, br, 1H, CO2H), 8.66 (s, 1H, H-2), 8.65 (s, 1H, H-8), 8.07 (d, 3 J H,H = 8.8 Hz, 2H, Ar), 7.05–7.20 (m, 12 H, Ar, MMT), 6.98 (d, 3 J H,H = 8.9 Hz, 2H, MMT), 6.66 (d, 3 J H,H = 9.0 Hz, 2H, MMT), 4.40 (m, 1H, H-β), 4.31 (m, 1H, H-β), 3.85 (s, 3H, anis.-OCH3), 3.67 (s, 3H, MMT–OCH3), 3.56 (m, 1H, H-α); 13C NMR (125 MHz, [D6]DMSO, 35°C) δ = 173.5, 162.5, 157.5, 157.4, 150.3, 147.9, 145.9, 145.8, 139.8, 137.1, 130.5, 129.5, 128.0, 127.9, 127.6, 126.1, 125.3, 113.6, 112.9, 69.5, 55.9, 55.4, 54.8, 46.6; MS (ESI) m/z 627.3 [M − H]−; HRMS (ESI): calcd. for C36H33N6O5: 629.2507; found 629.2513 [M + H]+.
(l)-N α -(4-Monomethoxytrityl)-β-[N4-(4-tert-butylbenzoyl)cytosin-1-yl]alanine (3)
Boc-β-[N4-(4-tert-butylbenzoyl)cytosine-1-yl]-alanine 10 (100 mg, 219 μmol) was dissolved in TFA (2 ml, 26.1 mmol) and stirred at room temperature for 2 h. TFA was removed in vacuo and the mixture repeatedly evaporated with toluene. After lyophilization from H2O, the residue was dissolved in DMF (40 ml). Triethylamine (88.3 mg, 876 μmol) and 4-monomethoxytritylchloride (136 mg, 0.44 mmol) in DMF (4 ml) were added and stirred at room temperature for 2 h. The solvent was removed in vacuo and the remaining oil was extracted three times with DCM/H2O (2:1, 200 ml). The organic phase was dried over Na2SO4 and concentrated in vacuo. Diethyl ether (3 ml) was added and the suspension was stirred at −28°C for 1 h. After centrifugation at −10°C and separation from diethyl ether a white solid was obtained. This procedure was repeated yielding amino acid 3 (33.8 mg, 25%). TLC R f 0.39 (AcOEt:MeOH = 4:1); \( \left[ \alpha \right]_{\text{D}}^{20} \) = –14.8° (c 0.12, CHCl3); IR (KBr): \( \tilde{\nu } \) = 3,432, 2,960, 1,691, 1,659, 1,488 cm−1; 1H NMR (500 MHz, [D6]DMSO, 35°C) δ = 11.18 (s, br, 1H, CO2H), 8.39 (d, 3 J H,H = 7.2 Hz, 1H, H-6), 7.98 (d, 3 J H,H = 8.4 Hz, 2H, Ar), 7.53 (d, 3 J H,H = 8.4 Hz, 2H, Ar), 7.40 (m, 1H, H-5), 7.10–7.30 (m, 12H, MMT), 6.76 (d, 3 J H,H = 8.9 Hz, 2H, MMT), 4.10 (m, 1H, H-β), 3.67–3.76 (m, 4H, H-β, MMT–OCH3), 3.66 (m, 1H, H-α), 1.30 (s, 9H, tBu); 13C NMR (125 MHz, [D6]DMSO, 35°C) δ = 173.6, 158.2, 157.5, 155.7, 151.4, 146.2, 145.8, 137.4, 129.6, 129.3, 128.3, 128.1, 128.0, 127.7, 126.1, 125.2, 113.0, 95.2, 69.5, 54.9, 54.7, 52.8, 34.7, 30.8; MS (ESI) m/z 629.3 [M − H]−; HRMS (ESI): calcd. for C38H39N4O5: 631.2915; found 631.2923 [M + H]+.
(l)-N α -(4-Methoxyphenyldiphenylmethyl)-β-[N2-(isobutyryl)-guanin-9-yl]-alanine (4)
TFA (6 ml, 78.3 mmol) was added to a suspension of Boc-β-[N2-(isobutyryl)guanine-9-yl]alanine 11 (200 mg, 490 μmol) in DCM (12 ml) at 0°C and the resulting solution was stirred at room temperature for 2 h. The solvent was removed in vacuo and the residue was dissolved in DMF (60 ml). Triethylamine (737 mg, 7.35 mmol) and 4-monomethoxytritylchloride (141 mg, 455 μmol) dissolved in DMF (5 ml) were added and the mixture stirred at room temperature for 1 h before the solvent was evaporated. The resulting oil was extracted five times with DCM/H2O (1:1, 200 ml), the organic phase was dried over Na2SO4 and the solvent removed. Diethyl ether (3 ml) was added and the suspension was stirred at −28°C for 1 h. After centrifugation at −10 °C and separation from diethyl ether a white solid was obtained. This procedure was repeated yielding amino acid 4 (87.3 mg, 31%). TLC R f 0.58 (AcOEt:MeOH = 1:1); \( \left[ \alpha \right]_{\text{D}}^{20} \) = +48.9° (c 0.1, MeOH); IR (KBr): \( \tilde{\nu } \) = 3,442, 2,930, 1,682, 1,607, 1,560 cm−1; 1H NMR (300 MHz, [D6]DMSO, 35°C) δ = 12.05 (s, br, 1H, CO2H), 11.53 (m, 1H, NH), 8.35 (s, 1H, H-8), 7.00–7.20 (m, 10H, MMT), 6.93 (d, 3 J H,H = 9.0 Hz, 2H, MMT), 6.62 (d, 3 J H,H = 9.1 Hz, 2H, MMT), 4.41 (m, 1H, H-β), 4.29 (m, 1H, H-β), 3.66 (s, 3H, OCH3), 3.56 (m, 1H, H-α), 2.76 (sept., 3 J H,H = 6.8 Hz, 1H, iBu), 1.10–1.20 (m, 6H, iBu-CH3); 13C NMR (75 MHz, CDCl3, 35°C) δ = 180.1, 172.2, 154.9,149.1, 147.8, 145.4, 145.5, 141.3, 136.9, 129.5, 127.9, 127.5, 126.2, 120.0, 112.8, 69.3, 56.0, 54.9, 45.8, 34.6, 18.9; MS (ESI) m/z 579.8 [M − H]−; HRMS (ESI): calcd. for C32H33N6O5: 581.2507; found 581.2510 [M + H]+.
Determination of enantiomeric excess
The alanyl nucleo amino acids (5.00 μmol, 1.00 eq.) were N-deprotected by TFA treatment. N-Boc-l-Ala-OSu (8.68 mg, 30.0 μmol, 6.00 eq.) was dissolved in 350 μl THF was added to the respective nucleo amino acid dissolved in water (175 μl) and NaHCO3 buffer (175 μl, pH 9, c = 1.2 mol/l). The mixture was kept for 5 h at room temperature before it was brought to pH 2.5 by addition of 10% TFA. The solid residue was filtered and 5 μl of the solution was used for HPLC analysis on a RP C18 column. The integration of the peak areas of both dipeptide diastereomers at 254 nm provided the enantiomeric excess.
Results and discussion
A molecular beacon composed of an alanyl-PNA stem and a DNA recognition part requires the preparation of alanyl-PNA/DNA/alanyl-PNA chimeras (Fig. 3a). They might be obtained either by ligation of two alanyl-PNA fragments to the 3′- and 5′-termini of the DNA single strand, respectively, (Ede et al. 1994; Arar et al. 1995; Vives and Lebleu 1997; Harrison and Balasubramanian 1998; McMinn and Greenberg 1999; Forget et al. 2001; Zatsepin et al. 2002; Ollivier et al. 2002) or by solid phase synthesis of the whole chimeric strand under DNA synthesis conditions (Bergmann and Bannwarth 1995; Truffert et al. 1996; Antopolsky and Azhayev 1999, 2000; Sakakura and Hayakawa 2000; Antopolsky et al. 2002; Stetsenko et al. 2002; Musumeci et al. 2004). Uhlmann and van Boom already reported the stepwise solid phase synthesis of aminoethylglycine-PNA/DNA-chimera (Fig. 3b) (Uhlmann et al. 1998, 1999; Vinyak 1999; Van der Laan et al. 1995, 1997) adapting the protecting group strategy for nucleo amino acids to DNA synthesis conditions. The protection of the nucleo amino acids needs to be compatible with the DNA phosphoramidite chemistry: the acid labile monomethoxytrityl (MMT) protection and the acyl protecting groups on the exocyclic amino functions of the nucleobases are suitable for this purpose (Stetsenko et al. 1996; Breipohl et al. 1997). Indeed, the MMT-protected monomers show good solubility in the polar organic solvents (e.g. DMF) used for the synthesis of the oligomers and the conditions for removing the MMT protecting group (3% trichloroacetic acid) are mild and do not cause depurination in the DNA portion of the chimeras. Moreover, in the peptide synthesis by MMT-chemistry every coupling can be monitored by measuring the absorbance of the released MMT+ ion (ε 478 = 65,000).

a Alanyl-PNA/DNA/alanyl-PNA-chimera and b Aminoethylglycine-PNA/DNA/aminoethylglycine-PNA-chimera as described by Uhlmann
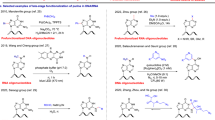
In this work, the enantioselective synthesis of MMT-protected nucleo amino acids 1–4 (Fig. 4) was provided for the first time by preparation of the four Boc-alanyl nucleo amino acids followed by an exchange of the terminal amino protecting group. Nevertheless, the need for acyl protection of the nucleobases afforded a reinvestigation of the Boc-amino acid synthesis with the respective nucleobase, thus, leading to the synthesis of the new Boc/acyl-protected nucleo amino acids (7, 10 and 11, Scheme 1).

MMT/acyl-protected nucleo amino acids for the solid phase synthesis of DNA/alanyl-PNA chimeras

Synthesis of Boc- and MMT-protected nucleo amino acids with acyl protected nucleobases
Synthesis of Boc-alanyl nucleo amino acids
Boc-protected alanyl nucleo amino acids are obtained by ring opening of Boc-l-serine lactone (5) (Arnold et al. 1985) with the respective nucleobase (Scheme 1) (Lohse et al. 1996; Diederichsen et al. 2005). Since thymine is the only nucleobase that does not require protection, nucleo amino acid 6 was obtained as described in literature (Lohse et al. 1996). Nα-Boc-β-[N6-(Anisoyl)adenine-9-yl]alanine (7) was provided in 32% yield by ring opening of Boc-l-serine lactone 5 with N6-anisoyl-adenine (8) in DMSO with DBU as a base. As known already for the reaction with the unprotected nucleobases, the yield of the serine lactone ring opening with DNA bases is quite low (22% in case of adenine) (Diederichsen et al. 2005) due to their low solubility, regioselectivity problems (N7 vs. N9 alkylation and SN2 vs. carbonyl attack on the serine lactone), polymerization of the serine lactone, and racemization. Nevertheless, despite extensive studies for improvement of this reaction, up to date this route turned out to be most efficient. The enantiomeric purity was investigated for nucleo amino acid 7 (92% ee) as well as for all other derivatives described herein by Boc-deprotection followed by dipeptide formation with the Boc-l-Ala-OSu active ester and HPLC analysis of the respective diastereomers. N4-(4-tert-Butylbenzoyl)cytosine (9) was also converted to the Nα-Boc-β-[N4-(4-tert-butylbenzoyl)cytosine-1-yl]alanine (10) in DMSO by the help of DBU with moderate yield (42%) but excellent enantiomeric purity (99% ee). Regarding synthesis of the (l)-Nα-Boc-β-[N2-(isobutyryl)guanin-9-yl]alanine (11), N2-(isobutyryl)guanine (12) was reacted with Boc-l-serine lactone (5) in DMSO and with DBU as base to yield the desired Boc-protected nucleo amino acid with N9-substituted isobutyryl-guanine 11 in a mixture with the N7 regioisomer (l)-Nα-Boc-β-[N2-(isobutyryl)guanin-7-yl]alanine. Subsequently, the mixture of N9 and N7 nucleo amino acids was separated by silica gel flash chromatography. The fractions containing the desired product, identified by comparison of the obtained 13C NMR data with those previously published for guanine-containing N9-regioisomers (Kovács and Schmél 2000; Rosenbohm et al. 2004), were combined and the solvent evaporated to dryness. In this way Nα-Boc-β-[N2-(isobutyryl)guanin-9-yl]alanine (11) was provided in 31% yield with an optical purity of 96% ee.
Synthesis of MMT-protected nucleo amino acids
The usual procedures for N-terminal MMT protection of amino acids (Mamos et al. 1988; Canle et al. 2000) require workup under basic conditions, which would lead to acyl deprotection in case of the nucleo amino acids. MMT-protected nucleo amino acids 1–4 were obtained starting from the respective Boc-derivatives 6, 7, 10 and 11 by Boc-deprotection with TFA and immediate tritylation of the free nucleo amino acids with monomethoxytrityl chloride in DMF in presence of triethylamine (Scheme 1). The desired protected monomers were directly obtained by precipitation from diethyl ether at −28°C. This straightforward route was chosen even because silica gel chromatography of the MMT-amino acids using acidic eluents for protonation of the carboxylate moiety causes the deprotection of the acid labile trityl protection.
Conclusion
The enantioselective preparation of all canonical alanyl nucleo amino acids with MMT-protected α-amino groups and acyl-protected exocyclic amino functions of the nucleobases was presented for the first time in this work. It was made possible by the synthesis of the respective N-Boc nucleo amino acids, carried out by adapting the synthetic routes to the Boc-protected alanyl amino acids in the case of the acyl-protected nucleobases, and, after Boc removal, their reprotection by the MMT group. These novel derivatives will be advantageous in the solid phase synthesis of alanyl-PNA/DNA chimeras, by conditions fully compatible with the standard phosphoramidite DNA synthesis strategy, which might serve as a tool for very sensitive DNA detection following the molecular beacon concept.
References
Antopolsky M, Azhayev A (1999) Stepwise solid phase synthesis of peptide–oligonucleotide conjugates on new solid supports. Helv Chim Acta 82:2130–2140
Antopolsky M, Azhayev A (2000) Stepwise solid phase synthesis of peptide–oligonucleotide phosphorothioate conjugates employing Fmoc peptide chemistry. Tetrahedron Lett 41:9113–9117
Antopolsky M, Azhayeva E, Tengvall U, Azhayev A (2002) A general method for the stepwise solid phase synthesis of peptide–oligonucleotide conjugates. Tetrahedron Lett 43:527–530
Arar K, Aubertin AM, Roche AC, Monsigny M, Mayer R (1995) Synthesis and antiviral activity of peptide–oligonucleotide conjugates prepared by using N-α-(bromoacetyl)peptides. Bioconjug Chem 6:573–577
Arnold LD, Kalantar TH, Vederas JC (1985) Conversion of serine to stereochemically pure β-substituted α-amino acids via β-lactones. J Am Chem Soc 107:7105–7109
Bergmann F, Bannwarth W (1995) Solid phase synthesis of directly linked peptide–oligodeoxynucleotide hybrids using standard synthesis protocols. Tetrahedron Lett 36:1839–1842
Breipohl G, Will DW, Peyman A, Uhlmann E (1997) Novel synthetic routes to PNA monomers and PNA–DNA linker molecules. Tetrahedron 53:14671–14686
Canle LM, Clegg W, Demirtas I, Elsegood MRJ, Maskill H (2000) Preparations, X-ray crystal structure determinations, and base strength measurements of substituted tritylamines. J Chem Soc Perkin Trans II, pp 85–92
Didenko VV (2002) In situ detection of DNA damage: methods and protocols. Humana Press, Totowa
Diederichsen U (1996) Pairing properties of alanyl peptide nucleic acids containing an amino acid backbone with alternating configuration. Angew Chem Int Ed Engl 35:445–448
Diederichsen U (1997a) Alanyl-PNA: evidence for linear band structures based on guanine-cytosine base pairs. Angew Chem Int Ed Engl 36:1886–1889
Diederichsen U (1997b) Alanyl-PNA oligomers: a new system for intercalation. Bioorg Med Chem Lett 7:1743–1746
Diederichsen U, Weicherding D (1999) Interactions of amino acid side chains with nucleobases in alanyl-PNA. Synlett S1:917–920
Diederichsen U, Weicherding D, Diezemann N (2005) Side chain homologation of alanyl peptide nucleic acids: pairing selectivity and stacking. Org Biomol Chem 3:1058–1066
Ede NJ, Tregear GW, Haralambidis J (1994) Routine preparation of thiol oligonucleotides: application to the synthesis of oligonucleotide–peptide hybrids. Bioconjug Chem 5:373–378
Ficht S, Mattes A, Seitz O (2004) Single-nucleotide-specific PNA-peptide ligation on synthetic and PCR DNA templates. J Am Chem Soc 126:9970–9981
Forget D, Boturyn D, Defrancq E, Lhomme J, Dumy P (2001) Highly efficient synthesis of peptide–oligonucleotide conjugates: chemoselective oxime and thiazolidine formation. Chem Eur J 7:3976–3984
Harrison JG, Balasubramanian S (1998) The synthesis and hybridisation properties of a library of oligonucleotide–peptide conjugates. Nucleic Acids Res 26:3136–3145
Howarth NM, Wakelin LPG (1997) Alpha-PNA: a novel peptide nucleic acid anologue of DNA. J Org Chem 62:5441–5450
Knemeyer JP, Sauer M (2000) Probes for detection of specific DNA sequences at the single-molecule level. Anal Chem 72:3717–3724
Kostrikis LG, Tyagi S, Mhlanga MM, Ho DD, Kramer FR (1998) Spectral genotyping of human alleles. Science 279:1228–1229
Kovács G, Schmél Z (2000) Fmoc/acyl protecting groups in the synthesis of polyamide (peptide) nucleic acid monomers. J Chem Soc Perkin Trans 1, pp 19–26
Kranaster R, Ketzer P, Marx A (2008) Mutant DNA polymerase for improved detection of single-nucleotide variations in microarrayed primer extension. Chembiochem 9:694–697
Küsel A, Zhang J, Alvarino Gil M, Stückl CA, Meyer-Klaucke W, Meyer F, Diederichsen U (2005) Metal binding within a peptide-based nucleobase stack with tuneable double strand topology. Eur J Inorg Chem 431:7–4324
Li JJ, Fang X, Schuster SM, Tan W (2000) Molecular beacons: a novel approach to detect protein–DNA interactions. Angew Chem Int Ed 39:1049–1052
Lohse P, Oberhauser B, Oberhauser-Hofbauer B, Baschang G, Eschenmoser A (1996) Chemie von alpha-aminonitrilen. XVII : Oligo(nukleoDipeptamidinium)-salze. Croat Chem Acta 69:535–562
Mamos P, Sanida C, Barlos K (1988) Preparation of N-tritylamino acids from N-trimethylsilylamino acid trimethylsilyl esters. Liebigs Ann Chem, pp 1083–1084
Matsuo T (1998) In situ visualization of messenger RNA for basic fibroblast growth factor in living cells. Biochim Biophys Acta 1379:178–184
McMinn DL, Greenberg MM (1999) Convergent solution-phase synthesis of a nucleopeptide using a protected oligonucleotide. Bioorg Med Chem 9:547–550
Musumeci D, Roviello GN, Valente M, Sapio R, Pedone C, Bucci EM (2004) New synthesis of PNA-3′DNA linker monomers, useful building blocks to obtain PNA/DNA chimeras. Biopolymers 76:535–542
Ollivier N, Olivier C, Gouyette C, Huynh-Dinh T, Gras-Masse H, Melnyk O (2002) Synthesis of oligonucleotide–peptide conjugates using hydrazone chemical ligation. Tetrahedron Lett 43:997–999
Pfeifer GP (1996) Technologies for detection of DNA damage and mutations. Plenum, New York
Rosenbohm C, Pedersen DS, Frieden M, Jensen FR, Arent S, Larsen S, Koch T (2004) LNA guanine and 2, 6-diaminopurine. Synthesis, characterization and hybridization properties of LNA 2, 6 diaminopurine containing oligonucleotides. Bioorg Med Chem 12:2385–2396
Sakakura A, Hayakawa Y (2000) A novel synthesis of oligonucleotide–peptide conjugates with a base-labile phosphate linker between the two components according to the allyl-protected phosphoramidite strategy. Tetrahedron 56:4427–4435
Socher E, Bethge L, Knoll A, Jungnick N, Herrmann A, Seitz O (2008) Low-noise stemless PNA beacons for sensitive DNA and RNA detection. Angew Chem Int Ed 47:9555–9559
Stetsenko DA, Lubyako EN, Potapov VK, Azhikina TL, Sverdlov ED (1996) New approach to solid phase synthesis of polyamide nucleic acids analogues (PNA) and PNA–DNA conjugates. Tetrahedron Lett 37:3571–3574
Stetsenko DA, Malakhov AD, Gait MJ (2002) Total stepwise solid-phase synthesis of oligonucleotide-(3′-N)-peptide conjugates. Org Lett 4:3259–3262
Tan W, Fang X, Li J, Liu X (2000) Molecular beacons: a novel DNA probe for nucleic acid and protein studies. Chem Eur J 6:1107–1111
Taylor GR (1997) Laboratory methods for the detection of mutations and polymorphisms in DNA. CRC, Boca Raton
Truffert JC, Asseline U, Brack A, Thuong NT (1996) Synthesis, purification and characterization of two peptide–oligonucleotide conjugates as new potential artificial nucleases. Tetrahedron 52:3005–3016
Tyagi S, Kramer FR (1996) Molecular beacons: probes that fluoresce upon hybridization. Nat Biotechnol 14:303–308
Tyagi S, Bratu D, Kramer FR (1998) Multicolor molecular beacons for allele discrimination. Nat Biotechnol 16:49–53
Uhlmann E, Peyman A, Breipohl G, Will DW (1998) PNA: synthetic polyamide nucleic acids with unusual binding properties. Angew Chem Int Ed 37:2797–2823
Uhlmann E, Greiner B, Breipohl G (1999) PNA/DNA chimeras. In: Nielsen PE, Egholm M (eds) Peptide nucleic acids, protocols and applications. Horizon Scientific Press, Norfolk, p 51
Valis L, Amann N, Wagenknecht H-A (2005) Detection of single base mismatches and abasic sites using phenanthridinium as an artificial DNA base and charge donor. Org Biomol Chem 3:36–38
Van der Laan AC, Meeuwenoord NJ, Kuyl-Yeheskiely E, Oosting RS, Brands R, van Boom JH (1995) Solid support synthesis of a PNA–DNA hybrid. Recl Trav Chim Pays Bas 114:295–297
Van der Laan AC, Brill AC, Kuimelis R, Kuyl-Yeheskiely E, van Boom JH (1997) A convenient automated solid-phase synthesis of PNA-(5′)-DNA-(3′)-PNA chimera. Tetrahedron Lett 38:2249–2252
Vinyak R (1999) Synthesis of PNA–DNA chimera by MMT chemistry. In: Nielsen PE, Egholm M (eds) Peptide nucleic acids, protocols and applications. Horizon Scientific Press, Norfolk, p 71
Vives E, Lebleu B (1997) Selective coupling of a highly basic peptide to an oligonucleotide. Tetrahedron Lett 38:1183–1186
Zatsepin TS, Stetsenko DA, Arzumanov AA, Romanova EA, Gait MJ, Oretskaya TS (2002) Synthesis of peptide–oligonucleotide conjugates with single and multiple peptides attached to 2′-aldehydes through thiazolidine, oxime, and hydrazine linkages. Bioconjug Chem 13:822–830
Acknowledgement
We are grateful for generous support by the Deutsche Forschungsgemeinschaft (Di 542/4-1).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s00726-009-0455-0
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Roviello, G.N., Gröschel, S., Pedone, C. et al. Synthesis of novel MMT/acyl-protected nucleo alanine monomers for the preparation of DNA/alanyl-PNA chimeras. Amino Acids 38, 1301–1309 (2010). https://doi.org/10.1007/s00726-009-0324-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-009-0324-x