
Abstract
Human coronavirus HKU1 (HCoV-HKU1) is a pathogen that causes acute respiratory tract infections in children and circulates worldwide. To investigate the molecular characteristics and genetic diversity of HCoV-HKU1 in China, a molecular epidemiological analysis based on complete genome sequences was performed. A total of 68 endemic-HCoV-positive samples were identified from 1358 enrolled patients during 2018, including four HCoV-229E, nine HCoV-OC43, 24 HCoV-NL63, and 31 HCoV-HKU1. The detection rate of endemic HCoVs was 5.01% during 2018, while for HCoV-HKU1, it was 2.28%. Eight complete genomic sequences of HCoV-HKU1 were obtained and compared to 41 reference genome sequences corresponding to genotypes A, B, and C, obtained from the GenBank databank. Of the eight HKU1 sequences, four belonged to genotype A and four belonged to genotype B. No genotype C strains were detected in this study. For genotype A, 18 variations in the S protein with respect to the reference sequence were present in more than 5% of the sequences, whereas for genotype B, this number was 25. Most of the amino acid changes occurred in the S1 subunit. No amino acid substitutions were found in the sites that are essential for interaction with neutralizing antibodies, while a 510T amino acid insertion was found in almost one third of genotype B sequences. About 82-83, 85-89, and 88-89 predicted N-glycosylation sites and 7-13, 6-8, and 9 predicted O-glycosylation sites were found among the sequences of genotype A, B, and C, respectively. Six conserved O-glycosylation sites were present in all of the genotype A sequences. Only genotype A and B strains were detected after 2005. The S protein exhibited relatively high diversity, with most of the amino acid changes occurring in the S1 subunit.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human coronaviruses (HCoVs) are enveloped, non-segmented, positive-sense single-stranded RNA viruses belonging to the family Coronaviridae, and they are the largest known RNA viruses, with a genome consisting of approximately 30,000 nucleotides with a single open reading frame (ORF) [4]. With the inclusion of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), seven pathogenic HCoVs have been identified. The subfamily Orthocoronavirinae is divided into four genera: Alpha-, Beta-, Gamma-, and Deltacoronavirus, with HCoVs belonging to the genera Alphacoronavirus (HCoV-229E and HCoV-NL63) and Betacoronavirus (HCoV-OC43, HCoV-HKU1, SARS-CoV, Middle East respiratory syndrome CoV [MERS-CoV] and SARS-CoV-2).
HCoV-229E, OC43, NL63, and HKU1 can infect the human respiratory tract and usually cause mild or moderate disease, but severe symptoms such as pneumonia are sometimes also reported in children and adults with comorbidities [20]. SARS-CoV-1, MERS-CoV, and SARS-CoV-2 can cause severe acute respiratory infection (SARI) and are emerging epidemic pathogens with significant morbidity, mortality, and economic impact [2]. HCoV-HKU1 is associated with acute respiratory tract infections (ARTI) and gastrointestinal disease, which are self-limiting. Fever, cough, runny nose, wheeze, hypoxia, seizures, and diarrhoea are the most common symptoms, while bronchiolitis, pneumonia, and asthmatic exacerbation have also been reported [6]. Children tend to be more susceptible to HKU1 infection than adults. The positive detection rate of HKU1 in patients ≤ 14 years old with ARTI is about 0.3% in Guangdong province in China [25].
HCoV-HKU1 was first discovered in adult pneumonia patients in Hong Kong in 2005 [22]. The HCoV-HKU1 genome ranges in length from 29,295 to 30,097 nt. Like those of other coronaviruses, the genome of HCoV-HKU1 has the characteristic gene order 5’-replicase ORF1ab (encoding the nonstructural polyproteins nsp1 to nsp16), hemagglutinin esterase (HE), spike (S), envelope (E), membrane (M), and nucleocapsid (N)-3’ [24]. The S glycoprotein consists of two subunits, S1 and S2. It mediates viral entry by first binding the host receptor via the S1 subunit and then fusing the viral and cellular membranes via the S2 subunit [14]. Based on phylogenetic analysis of the RNA-dependent RNA polymerase (RdRp), S, and N genes, HKU1 isolates are separated into three genotypes, named A, B, and C, with intergenotypic homologous recombination occurring between their genomes [23].
Molecular characterization of the viral genome is important for understanding the molecular epidemiology of HKU1 in China and obtaining data necessary for the prevention and control of disease. However, there is a lack of genome sequences of HCoV-HKU1 strains. The present multicenter study was performed to investigate the epidemiology and genetic characteristics of HKU1 in mainland China during 2018.
Materials and methods
Sample collection and HKU1 detection
Respiratory samples were collected in 2018 from patients hospitalized with community-acquired pneumonia (CAP) in a multicenter study in China that involved five hospitals from five provinces or municipalities (Beijing, Guangdong, Zhejiang, Guizhou, and Ningxia). The eligible participants were under 18 years of age. Nasopharyngeal swab specimens were collected on admission and stored at −80℃. Total DNA and RNA were extracted by using a QIAamp MinElute Virus Spin Kit (QIAGEN, Hilden, Germany). HCoV-229E, OC43, NL63, and HKU1 were detected using a multiplex real-time PCR assay (Multiplex Real-Time PCR Diagnostic Kit for Rapid Detection of 15 respiratory viruses; XABT, Beijing, China).
Sequence determination of HKU1 genomes
For HKU1-positive samples, the complete genome sequences were amplified in segments by overlapping PCR, using a SuperScript™ III One-Step RT-PCR System (Invitrogen, Carlsbad CA, USA, catalog no. 12574018). All of the amplicons were pooled and then barcoded and sequenced using next-generation sequencing platforms. The primer pairs used for amplification and sequencing are shown in Supplementary Table S1. These genome sequences were submitted to the GenBank database with the accession numbers ON128609-ON128616.
Genetic characterization and phylogenetic analysis
HKU1 genome sequences retrieved from the GenBank database (Supplementary Table S2) were analyzed together with the sequences from this study. MEGA software (Version 6.06, Sudhir Kumar, Arizona State University, Tempe, AZ, USA) was used for multiple sequence alignments, pairwise distance measurements, and phylogenetic analysis. Sequences were compared and genetic diversity was analyzed using BioEdit software (Version 7.2.5). A phylogenetic tree was constructed by the neighbor-joining method, using the Kimura 2-parameter model. The reliability of phylogenetic inference was estimated by the bootstrap method with 1000 replicates, with values over 75% shown in the tree.
Glycosylation analysis
N-linked and O-linked amino acid glycosylation sites in the S protein of HKU1 were predicted using the NetNGlyc 1.0 (https://services.healthtech.dtu.dk/service.php?NetNGlyc-1.0) and NetOGlyc 4.0 (https://services.healthtech.dtu.dk/service.php?NetOGlyc-4.0) server, respectively. The sequence N-X-S/T (where X is not Pro) was defined as a potential N-linked glycosylation site, and S andT residues were identified as potential O-linked sugar acceptors. Only sites with scores higher than 0.5 were considered likely to be glycosylated.
Results
Detection of HCoVs in nasopharyngeal swab specimens
A total of 68 (5.01%, 68/1358) endemic-HCoV-positive samples were identified from 1358 enrolled patients during 2018. The number of samples containing HCoV-229E, OC43, NL63, and HKU1 was 4 (0.29%), 9 (0.66%), 24 (1.76%), and 31 (2.28%), respectively. The ages of these children ranged from 1 month to 9.5 years, with a median age of 1 year. The male-to-female ratio was 2.09:1 (46 boys and 22 girls).
Other respiratory viruses were also detected in 46 (67.65%) HCoV-positive samples, including bocavirus (HBoV) (17, 25.00%), enterovirus/rhinovirus (EV/HRV) (12, 17.65%), human respiratory syncytial virus (HRSV) (6, 8.82%), adenovirus (HAdV) (5, 7.35%), parainfluenza virus type 3 (HPIV3) (5, 7.35%), and influenza virus type A (Flu A) (3, 4.41%).
For the patients infected with HKU1, the main clinical manifestations were pneumonia, lobular pneumonia, or bronchitis. The ages of these children ranged from 1 month to 6 years, with a median age of 11 months. The male-to-female ratio was 1.58:1.
Phylogenetic analysis of complete genome and S, RdRp, and N gene sequences
In total, eight complete genome sequences of HKU1 were obtained in this study and submitted to the GenBank database. Phylogenetic analysis was performed with reference sequences retrieved from GenBank.
The phylogenetic tree based on complete genomic sequences formed three clusters, corresponding to genotypes A, B, and C, respectively. The mean genetic distances were 0.0029, 0.0030, and 0.0006 within genotype A, genotype B, and genotype C, respectively (Fig. 1). The mean genetic distances were 0.0477, 0.0367, and 0.0192 between genotype A and B, A and C, and B and C, respectively. The pairwise nucleotide sequence identities were 97.1-99.9%, 98.5–99.9%, and 99.4–99.9% within genotype A, B, and C, and 92.4-94.8%, 93.7–95.9%, and 97.8–98.1% between genotypes A and B, A and C, B and C, respectively. A comprehensive comparison of nucleotide and predicted amino acid sequences based on each gene was also performed (Supplementary Table S3). The sequences of different protein-encoding regions were highly conserved within each genotype. Between genotypes A and B, A and C, the structural-protein-encoding genes HE, S, and E exhibited much more diversity, while the other gene regions were relatively conserved (Supplementary Table S3).

Phylogenetic dendrogram based on the complete genome sequences (A), S (B), RdRp (C), and N (D) gene sequences of HCoV-HKU1 isolates from this study and reference sequences from GenBank. The topology was inferred using the neighbor-joining method, using MEGA 7.0 software. Bootstrap values (percentage of 1000 pseudoreplicate datasets) over 75% are shown at the nodes of the trees. The evolutionary distances were computed using the Kimura 2-parameter method. The scale bars represent substitutions per base pair per indicated horizontal distance. Red filled circles indicate the strains from this study, and strains colored in green are of genotype C.
Of the eight sequences determined in this study, BCH18024, WZ17198, WZ17244, and GY18102, collected from Beijing, Zhejiang, and Guizhou, belonged to genotype A, while GZFE18139, GZFE18147, GZFE18183, and GZFE18186 from Guangdong province belonged to genotype B. The four sequences of genotype A in this study were clustered together, with a mean genetic distance of 0.0013, while the mean genetic distance among the four genotype B sequences was 0.0006. The sequences of genotype C were all from Hong Kong, China, during 2003 and 2004. The phylogenetic trees based on the RdRp gene, N gene, and S gene stratified into two clusters; while the strains of genotype C clustered together with genotype B in the S and N gene trees, together with genotype A in the RdRp gene tree (Fig. 1). Separate phylogenetic trees based on the nsp, HE, E, M, and N2 genes were also constructed (Supplementary Fig. S1). Consistent with the dendrogram based on RdRp gene sequences, most of the trees based on the nsp genes showed that the genotype C isolates clustered together with the genotype A isolates. However, the strains of genotype C clustered together with genotype B in the trees based on the M, HE, E, and N2 gene sequences, which is similar to the results obtained with the S and N genes.
Variation in the deduced amino acid sequence of the S protein
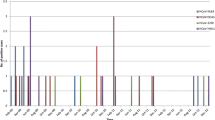
The coronavirus S glycoprotein is important for interaction between the virus and the host cell. As a class I viral fusion protein, the S protein is also an important target for vaccine and neutralizing antibody development. The deduced S protein length of HKU1 genotype A, genotype B, and genotype C was 1356, 1352/1353, and 1352 amino acids, respectively. The S glycoprotein consists of two subunits S1 (aa 14-757) and S2 (aa 901-1356), with S1 mediating receptor recognition and binding and S2 mediating membrane fusion and virus entry. Compared with the reference sequence (HKU1 genotype A, GenBank accession number AY597011), 24 amino acid variations were detected in the S protein of the genotype A isolates, excluding unique differences found in individual sequences. Eighteen of these variations were present in more than 5% of the sequences, 13 of which were present in more than 10% of the sequences (Fig. 2). Most of the amino acid changes occurred in the S1 subunit. Six amino acid substitutions were detected in all four genotype A sequences of this study when compared to the reference sequence (AY597011): L28K, S70A, V387I, K478N, H513R, and V567A. The C-terminal domain (CTD) of the HKU1 S1 subunit contains the receptor-binding domain (RBD) and the epitopes for neutralizing antibodies. The amino acids V509, L510, D511, H512, W515, and R517 are known to be important for neutralizing antibody interactions, but no amino acid substitutions were found at these sites.

Summary of amino acid changes and frequencies in the S gene of genotype A (n = 3), and B (n = 193) in comparison to reference sequences. The earliest sequence of each genotype was used as a reference (A: AY597011; B: AY884001). Only changes with the frequencies greater than 5% are shown.
The S proteins of genotypes B and C are almost identical. In comparison with the reference sequence (AY884001), 25 amino acid variations were present in more than 5% of the genotype B sequences, 19 of which were present in more than 10% of the sequences. Like in genotype A, most of the amino acid changes occurred in the S1 subunit. The amino acid insertion 510T was found in almost one third of the genotype B sequences (Fig. 2).
Predicted glycosylation sites in the S protein
The N-X-S/T sequons, where X is any amino acid except proline, were identified as potential N-glycosylation sites. About 82-83, 85-89, and 88-89 predicted N-glycosylation sites were found among the sequences of genotype A, B, and C, respectively. Eighty-three predicted N-glycosylation sites were identified among these four sequences of genotype A in this study. For the four genotype B sequences in this study, 86 predicted potential N-glycosylation sites were identified.
The S protein also contains a number of serine and threonine residues, which are potential O-glycosylation sites. About 7-13, 6-8, and 9 predicted O-glycosylation sites were identified in the S proteins of genotype A, B, and C, respectively. Six conserved O-glycosylation sites (aa 484, 487, 492, 761, 870, and 1246) were found in all of the sequences of genotype A, while there was an extra conserved site (S751) in all of the Chinese sequences. There were four conserved O-glycosylation sites (484, 492, 529, and 759) in all of the genotype B sequences, while two extra conserved site (527 and 760) were identified in all of the Chinese sequences (G-scores: 0.5 to 0.8).
Discussion
HCoVs are important pathogens with global distribution that cause human respiratory infections. Within the genus Betacoronavirus, interspecies transmission is a common phenomenon, leading to the emergence of new pathogens [10, 15]. The characteristics of cross-species transmission and rapid evolution have contributed to the emergence of new HCoVs strains or variants, some of which cause severe disease in humans and place a tremendous burden on society, such as SARS-CoV, MERS-CoV, and SARS-CoV-2 [3, 8, 11, 17]. The rapid and continuous evolution of SARS-CoV-2 has resulted in many variants with increased transmissibility [12]. HKU1, which, like HCoV-OC43, SARS-CoV, MERS-CoV, and SARS-CoV-2, belongs to the genus Betacoronavirus, was first reported in Hong Kong in 2005 and then circulated endemically [21]. Unlike SARS-CoV, MERS-CoV, and SARS-CoV-2, HKU1-associated ARTI is usually mild and self-limiting. HCoV-HKU1 is relatively conserved, without novel genotypes or variants being reported. However, we still need to pay attention to this virus, especially its genomic characteristics and variations.
In this study, 1358 children hospitalized in China during 2018 with CAP were enrolled. Sixty-eight HCoV-positive samples were identified, with a detection rate of 5.01%, while the rates of HCoV-229E, OC43, NL63, and HKU1 were 0.29%, 0.66%, 1.76%, and 2.28%, respectively. These results are roughly consistent with those of other studies [16, 25].
Co-detection of HCoVs with other respiratory viruses is frequent in children with ALRTI, with HBoV, HRSV, EV, HPIV, and HAdV being the most frequently co-detected viruses [16, 25, 29]. The clinical implications of such coinfections need further analysis of clinical data. It has been reported that HRSV and HRV coinfection with HCoVs did not exacerbate the disease presentation in patients [5, 7]. In the present study, the most commonly co-detected viruses from HCoV-positive samples were HBoV (25.00%), EV/HRV (17.65%), HRSV (8.82%), HAdV (7.35%), and HPIV3 (7.35%). These results are roughly consistent with previously studies from around the world [5, 16, 25, 29].
There are three known genotypes of HCoV-HKU1 A, B, and C, with genotype C having arisen from recombination of genotypes A and B [23]. No new genotypes or recombination events were identified in this study. The results showed that the gene sequences were conserved within the corresponding genotypes. In contrast, in the case of HCoV-OC43, novel genotypes arise continually by recombination, and 11 genotypes have been reported so far [26, 28]. SARS-CoV-2 is also evolving rapidly, and the Omicron variant has become the dominant lineage, showing an incomparable transmission speed [27].
The S glycoprotein of HCoV-HKU1 is a class I viral fusion protein that affects the tissue tropism, host range, and virulence of the virus. The S glycoprotein contains two subunits, S1 and S2, which, in the spike precursor, are separated by a protease-sensitive amino acid sequence. The S1 subunit mediates receptor recognition and binding, while the S2 subunit mediates membrane fusion and virus entry [19] . The S protein is also an important target for vaccine and neutralizing antibody development [13, 18]. Amino acid changes in the S protein can result in changes in virulence or immune escape, and this brings challenges for virus prevention and control as well as vaccine development. It is therefore of great importance to monitor variations in the S protein. Among the genotype A sequences, 18 amino acid variations in the S protein were present in more than 5% of the sequences, 13 of which were present in more than 10% of the sequences. Among the genotype B sequences, 25 amino acid variations in the S protein were present in more than 5% of the sequences, 19 of which were present in more than 10% of the sequences. Most of the amino acid changes occurred in the S1 subunit, which contains the RBD, but no amino acid substitutions were identified at V509, L510, D511, H512, W515, or R517, which are important for neutralizing antibody interactions in genotype A. Interestingly, an amino acid insertion (510T) was found in almost one third of the genotype B sequences. Further work is needed to investigate the effect of this insertion on the virus.
N-linked glycans of the S protein play important roles in virus recognition, binding, affinity, infectivity, antigenicity, and immune evasion. Mutation of important N-glycosylation sites can affect the stability of S protein, the sensitivity of the virus to neutralizing antibodies, and the infectivity of the virus [9]. In this study, about 82-83, 85-89, and 88-89 predicted N-glycosylation sites were identified in the S proteins of genotype A, B, and C, respectively. The S protein of HKU1 has a high N-glycosylation density, with a small amount of variation among the strains. In addition to the N-glycosylation sites, many potential O-glycosylation sites were also identified; however, their functions remain unclear. Some of the predicted O-glycosylation sites located close to N-glycosylation sites may have complementary functions in immune shielding [1, 9]. In this study, 7-13, 6-8, and 9 predicted O-glycosylation sites were identified in the protein S of genotype A, B, and C, respectively. In the S proteins of genotype A, six conserved O-glycosylation sites were present in all of the sequences, while some were present in specific strains from different regions or time periods.
In conclusion, this is a multicenter molecular epidemiologic study of HCoV-HKU1 in China during 2018. Only genotypes A and B have been detected after 2005. The S gene sequences were relatively conserved within each genotype, which formed defined clusters according to genotypes. However, the S protein still exhibits relatively high diversity, with most of the amino acid sequence variability in the S1 subunit. No amino acid substitutions were found in sites that are essential for the neutralizing antibody interactions. No new recombinant strains have been reported since 2005, in contrast to HCoV-OC43, another betacoronavirus for which new genotypes are generated frequently by recombination. Continued surveillance and genome sequencing is needed to monitor the genetic variation of coronaviruses.
References
Bagdonaite I, Thompson AJ, Wang X, Sogaard M, Fougeroux C, Frank M, Diedrich JK, Yates JR, Salanti A, Vakhrushev SY, Paulson JC, Wandall HH (2021) Site-specific O-glycosylation analysis of SARS-CoV-2 spike protein produced in insect and human cells. Viruses 13:551
Bonilla-Aldana DK, Quintero-Rada K, Montoya-Posada JP, Ramirez-Ocampo S, Paniz-Mondolfi A, Rabaan AA, Sah R, Rodriguez-Morales AJ (2020) SARS-CoV, MERS-CoV and now the 2019-novel CoV: Have we investigated enough about coronaviruses?—A bibliometric analysis. Travel Med Infect Dis 33:101566
Chan-Yeung M, Xu RH (2003) SARS: epidemiology. Respirology 8(Suppl):S9-14
Coleman CM, Frieman MB (2014) Coronaviruses: important emerging human pathogens. J Virol 88:5209–5212
Dominguez SR, Robinson CC, Holmes KV (2009) Detection of four human coronaviruses in respiratory infections in children: a one-year study in Colorado. J Med Virol 81:1597–1604
Dominguez SR, Shrivastava S, Berglund A, Qian Z, Goes LGB, Halpin RA, Fedorova N, Ransier A, Weston PA, Durigon EL, Jerez JA, Robinson CC, Town CD, Holmes KV (2014) Isolation, propagation, genome analysis and epidemiology of HKU1 betacoronaviruses. J Gen Virol 95:836–848
Gaunt ER, Hardie A, Claas EC, Simmonds P, Templeton KE (2010) Epidemiology and clinical presentations of the four human coronaviruses 229E, HKU1, NL63, and OC43 detected over 3 years using a novel multiplex real-time PCR method. J Clin Microbiol 48:2940–2947
Giovanetti M, Benedetti F, Campisi G, Ciccozzi A, Fabris S, Ceccarelli G, Tambone V, Caruso A, Angeletti S, Zella D, Ciccozzi M (2021) Evolution patterns of SARS-CoV-2: Snapshot on its genome variants. Biochem Biophys Res Commun 538:88–91
Gong Y, Qin S, Dai L, Tian Z (2021) The glycosylation in SARS-CoV-2 and its receptor ACE2. Signal Transduct Target Ther 6:396
Gossner C, Danielson N, Gervelmeyer A, Berthe F, Faye B, Kaasik Aaslav K, Adlhoch C, Zeller H, Penttinen P, Coulombier D (2016) Human-dromedary camel interactions and the risk of acquiring zoonotic middle east respiratory syndrome coronavirus infection. Zoonoses Public Health 63:1–9
Hu B, Guo H, Zhou P, Shi ZL (2021) Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol 19:141–154
Karim SSA, Karim QA (2021) Omicron SARS-CoV-2 variant: a new chapter in the COVID-19 pandemic. Lancet 398:2126–2128
Kirchdoerfer RN, Cottrell CA, Wang N, Pallesen J, Yassine HM, Turner HL, Corbett KS, Graham BS, McLellan JS, Ward AB (2016) Pre-fusion structure of a human coronavirus spike protein. Nature 531:118–121
Li F (2016) Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol 3:237–261
Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang H, Crameri G, Hu Z, Zhang H, Zhang J, McEachern J, Field H, Daszak P, Eaton BT, Zhang S, Wang LF (2005) Bats are natural reservoirs of SARS-like coronaviruses. Science 310:676–679
Matoba Y, Abiko C, Ikeda T, Aoki Y, Suzuki Y, Yahagi K, Matsuzaki Y, Itagaki T, Katsushima F, Katsushima Y, Mizuta K (2015) Detection of the human coronavirus 229E, HKU1, NL63, and OC43 between 2010 and 2013 in Yamagata, Japan. Jpn J Infect Dis 68:138–141
Memish ZA, Perlman S, Van Kerkhove MD, Zumla A (2020) Middle East respiratory syndrome. Lancet 395:1063–1077
Ou X, Guan H, Qin B, Mu Z, Wojdyla JA, Wang M, Dominguez SR, Qian Z, Cui S (2017) Crystal structure of the receptor binding domain of the spike glycoprotein of human betacoronavirus HKU1. Nat Commun 8:15216
Qian Z, Ou X, Goes LG, Osborne C, Castano A, Holmes KV, Dominguez SR (2015) Identification of the receptor-binding domain of the spike glycoprotein of human betacoronavirus HKU1. J Virol 89:8816–8827
Veiga A, Martins LG, Riediger I, Mazetto A, Debur MDC, Gregianini TS (2020) More than just a common cold: endemic coronaviruses OC43, HKU1, NL63, and 229E associated with severe acute respiratory infection and fatality cases among healthy adults. J Med Virol 93:1002–1007
Woo PC, Lau SK, Chu CM, Chan KH, Tsoi HW, Huang Y, Wong BH, Poon RW, Cai JJ, Luk WK, Poon LL, Wong SS, Guan Y, Peiris JS, Yuen KY (2005) Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol 79:884–895
Woo PC, Lau SK, Tsoi HW, Huang Y, Poon RW, Chu CM, Lee RA, Luk WK, Wong GK, Wong BH, Cheng VC, Tang BS, Wu AK, Yung RW, Chen H, Guan Y, Chan KH, Yuen KY (2005) Clinical and molecular epidemiological features of coronavirus HKU1-associated community-acquired pneumonia. J Infect Dis 192:1898–1907
Woo PC, Lau SK, Yip CC, Huang Y, Tsoi HW, Chan KH, Yuen KY (2006) Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1. J Virol 80:7136–7145
Woo PC, Lau SK, Yip CC, Huang Y, Yuen KY (2009) More and more coronaviruses: human coronavirus HKU1. Viruses 1:57–71
Zeng ZQ, Chen DH, Tan WP, Qiu SY, Xu D, Liang HX, Chen MX, Li X, Lin ZS, Liu WK, Zhou R (2018) Epidemiology and clinical characteristics of human coronaviruses OC43, 229E, NL63, and HKU1: a study of hospitalized children with acute respiratory tract infection in Guangzhou, China. Eur J Clin Microbiol Infect Dis 37:363–369
Zhang Z, Liu W, Zhang S, Wei P, Zhang L, Chen D, Qiu S, Li X, Zhao J, Shi Y, Zhou R, Wang Y, Zhao J (2022) Two novel human coronavirus OC43 genotypes circulating in hospitalized children with pneumonia in China. Emerg Microbes Infect 11:168–171
Zhao Y, Huang J, Zhang L, Lian X, Wang D (2022) Is the Omicron variant of SARS-CoV-2 coming to an end? Innovation (Camb) 3:100240
Zhu Y, Li C, Chen L, Xu B, Zhou Y, Cao L, Shang Y, Fu Z, Chen A, Deng L, Bao Y, Sun Y, Ning L, Liu C, Yin J, Xie Z, Shen K (2018) A novel human coronavirus OC43 genotype detected in mainland China. Emerg Microbes Infect 7:173
Zhu Y, Xu B, Li C, Chen Z, Cao L, Fu Z, Shang Y, Chen A, Deng L, Bao Y, Sun Y, Ning L, Yu S, Gu F, Liu C, Yin J, Shen A, Xie Z, Shen K (2021) A multicenter study of viral aetiology of community-acquired pneumonia in hospitalized children in Chinese mainland. Virol Sin 36:1543–1553
Funding
This work was supported by the National Science and Technology Major Projects (grant number 2017ZX10104001-005-010, 2017ZX10103004-004), and the CAMS Innovation Fund for Medical Sciences (CIFMS) (grant number 2019-I2M-5-026). These sponsors had no role in the study design, data analysis, manuscript preparation, or decision to publish.
Author information
Authors and Affiliations
Contributions
X.C. and Z.X. conceived and designed the experiments; X.C., Y.Z., and Q.L. performed the experiments; X.C., Y.Z., G.L., C.L., R.J., L.L., B.X. L.G., and J.Y. contributed specimens and analyzed the data; and X.C. and Z.X. contributed to the writing of the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Animal and human rights statement
This study was approved by the Medical Ethics Committee of the Beijing Children’s Hospital, affiliated to Capital Medical University, and informed consent was obtained. All procedures were conducted according to the guidelines (Ethics Number 2017-k-15).
Additional information
Handling Editor: Pablo Pineyro.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
705_2022_5541_MOESM1_ESM.pdf
Supplementary Fig. S1 Phylogenetic dendrogram based on the HCoV-HKU1 nsp1 (A), nsp2 (B), nsp3 (C), nsp4 (D), nsp5 (E), nsp6 (F), nsp7 (G), nsp10 (H), nsp11 (I), nsp12 (J), nsp13 (K), M (L), HE (M), E (N), and N2 (O) gene sequences. The topology was inferred using the neighbor-joining method in MGEA 7.0 software. Evolutionary distances were computed using the Kimura 2-parameter method. Bootstrap values (percentage of 1000 pseudoreplicate datasets) over 75% are shown at the nodes of the trees. The scale bars represent substitutions per base pair per indicated horizontal distance. Red filled circles indicate the strains from this study, while green filled triangles are the strains of genotype C
Rights and permissions
About this article

Cite this article
Chen, X., Zhu, Y., Li, Q. et al. Genetic characteristics of human coronavirus HKU1 in mainland China during 2018. Arch Virol 167, 2173–2180 (2022). https://doi.org/10.1007/s00705-022-05541-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-022-05541-4