
Abstract
The global emergence and re-emergence of arthropod-borne viruses (arboviruses) over the past four decades have become a public health crisis of international concern, especially in tropical and subtropical countries. A limited number of vaccines against arboviruses are available for use in humans; therefore, there is an urgent need to develop antiviral compounds. Snake venoms are rich sources of bioactive compounds with potential for antiviral prospection. The major component of Crotalus durissus terrificus venom is a heterodimeric complex called crotoxin, which is constituted by an inactive peptide (crotapotin) and a phospholipase A2 (PLA2-CB). We showed previously the antiviral effect of PLA2-CB against dengue virus, yellow fever virus and other enveloped viruses. The aims of this study were to express two PLA2-CB isoforms in a prokaryotic system and to evaluate their virucidal effects. The sequences encoding the PLA2-CB isoforms were optimized and cloned into a plasmid vector (pG21a) for recombinant protein expression. The recombinant proteins were expressed in the E. coli BL21(DE3) strain as insoluble inclusion bodies; therefore, the purification was performed under denaturing conditions, using urea for protein solubilization. The solubilized proteins were applied to a nickel affinity chromatography matrix for binding. The immobilized recombinant proteins were subjected to an innovative protein refolding step, which consisted of the application of a decreasing linear gradient of urea and dithiothreitol (DTT) concentrations in combination with the detergent 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate (CHAPS) as a protein stabilizer. The refolded recombinant proteins showed phospholipase activity and virucidal effects against chikungunya virus, dengue virus, yellow fever virus and Zika virus.
Similar content being viewed by others


Introduction
The global emergence and re-emergence of arthropod-borne viruses (arboviruses) over the past four decades have become a public health crisis of international concern, especially in tropical and subtropical countries [40, 45]. In Brazil, the most frequent arboviruses are dengue virus (DENV), Zika virus (ZIKV), and yellow fever virus (YFV) of the genus Flavivirus, family Flaviviridae, chikungunya virus (CHIKV) of the genus Alphavirus, family Togaviridae, and Oropouche virus (OROV) of the genus Orthobunyavirus, family Peribunyaviridae, although several sporadic cases of disease caused by other arboviruses have also been reported [16, 17]. The new era of dengue epidemics in Brazil started in 1986 with the introduction of dengue virus serotype 1 (DENV-1), followed by the introduction of DENV-2 (1990), DENV-3 (2000) and DENV-4 (2008). More than 10,000,000 dengue cases and thousands of death occurred in this period. Infection with OROV can lead to Oropouche fever, which is an acute febrile illness similar to dengue with symptoms such as fever, headache, muscle and joint pain, and skin rash. The emergence and re-emergence of OROV fever in Central and South America have resulted in more than 30 epidemics in Brazil, Peru, Panama, and Trinidad and Tobago, the majority being reported in Brazil. OROV is the second most frequent arbovirus in Brazil after DENV, with considerable social and economic impact. It is estimated that over half a million people have been infected by OROV in over 60 years [37]. The first epidemic of CHIKV occurred in Bahia State in the Northeastern region of Brazil. The virus spread to several regions of the country and was responsible for more than 500,000 cases of chikungunya fever, which is an acute febrile illness affecting the joints [2]. The main concern about CHIKV is the possibility of causing a chronic disease with joint pain and deformities. Mayaro virus (MAYV) is another alphavirus that is endemic in the Amazon and the Central Plateau of Brazil. Infection with MAYV can lead to an acute febrile illness associated with prolonged and painful joint inflammation and swelling. MAYV usually circulates in a sylvatic cycle of forest mosquitoes and vertebrates. Sporadic cases of MAYV infection occur every year, but this virus represents a serious threat to humans because of its ability to replicate in Aedes aegypti mosquitoes [23]. ZIKV infections were initially reported in February 2015 in Bahia State, and the virus then spread across the country. By October 2018, the public health surveillance system had reported more than 230,000 cases of Zika. A large number of Guillain-Barré syndrome and fetal microcephaly cases have been reported in the country associated with ZIKV infection [24].
There is no antiviral drug licensed for the treatment of arbovirus infections. In addition, although a limited number of vaccines to prevent arbovirus infection are available, most of them are live attenuated vaccines that are mainly administered to people living in or traveling to endemic areas and are contraindicated during pregnancy or immunosuppression [9]. Thus, the vaccines are not sufficient to prevent the diseases caused by arboviruses. For example, although a licensed vaccine against YFV (genus Flavivirus, family Flaviviridae) is available, outbreaks of yellow fever have become increasingly common in the Americas and Africa, where the virus is endemic [9]. Therefore, there is an urgent need for the development of antiviral compounds to combat these viruses.
Snake venoms are a mixture of bioactive compounds with different biochemical, functional and structural properties and constitute interesting molecular models for the prospection of therapeutic agents [11, 22, 33]. Among the toxins found in snake venoms, the phospholipases A2 (PLA2s - EC 3.1.1.4) are important enzymes that are involved in the intoxication processes. The PLA2s can be found in several organisms and represent a class of versatile enzymes based on their functions, locations, amino acid sequences, structures, and mechanisms of action. Generally, PLA2s catalyze the hydrolysis of the sn-2 position (according to phospholipid carbon nomenclature) of phospholipids and release free fatty acids and lysophospholipids as products [10]. The PLA2s are classified into six main types: secreted (sPLA2), cytosolic (cPLA2), calcium-independent (iPLA2), platelet-activating factor acetylhydrolase (PAF-AH), lysosomal (LPLA2), and adipose-specific (AdPLA). The snake venom PLA2s (svPLA2) belong to groups IA and IIA of sPLA2; they have six to eight disulfide bonds, are calcium-dependent and are 13-18 kDa in size [29, 32]. The svPLA2s have been widely studied and have shown potential for biomedical applications as antiviral [27, 28, 35], antibacterial [38], anti-parasitic [5, 30], anti-platelet [41], hypotensive [41], and antitumoral [34] agents.
Crotoxin, which is a potent neurotoxin and the main component of the venom of snakes of the genus Crotalus, was the first toxin to be isolated and crystallized [42]. Crotoxin is a reversible heterodimeric protein complex consisting of crotapotin (CA; crotoxin A or acid component), which is an acidic non-toxic polypeptide without enzymatic activity, and a basic PLA2 (CB; crotoxin B or basic component), which is responsible for the enzymatic activity and toxicity exerted by the complex [19, 36]. Crotapotin acts as a chaperone of PLA2-CB, thereby preventing binding to non-specific sites and enhancing the enzymatic activity. The PLA2-CB/CA complex dissociates after binding to the target; after which the biological activities attributed to crotoxin result from the action of PLA2-CB [12, 31]. Two PLA2-CB isoforms (molecular weights of 14.180 Da [PLA2-CB1] and 14.244 Da [PLA2-CB2]) have been identified previously in the venom of Crotalus durissus terrificus [6]. We previously showed that PLA2-CB isolated from Crotalus durissus terrificus venom exerts antiviral effects against DENV and other enveloped viruses by direct action on the viral envelopes [28]. We also showed that the enzymatic activity is important for the antiviral effect exerted by PLA2-CB [35].
In this study, we describe the expression of two recombinant PLA2-CB isoforms in a prokaryotic system and their virucidal activity against dengue virus type 2, CHIKV, YFV, and ZIKV.
Materials and methods
Ethics statement
All animal experiments were performed according to the guidelines of the Brazilian College of Animal Experimentation and were approved by the Ethical Committee on Animal Experimentation of the Campus of Ribeirao Preto, University of Sao Paulo (CEUA/USP-RP, permit no. 12.1.1854.53.8).
Preparation of native PLA2-CB
Native PLA2-CB was purified from the lyophilized crude venom of Crotalus durissus terrificus as described previously [19]. Crotalus durissus terrificus venom was obtained from the serpentarium of the Ribeirao Preto Medical School, University of Sao Paulo (IBAMA authorization: 1/35/1998/000846-1).
Viruses
CHIKV (strain S27), DENV-2 (strain NGC), YFV (strain 17D) and ZIKV (isolated from a patient) obtained from the virus repository of the Virology Research Center, Ribeirao Preto Medical School, University of Sao Paulo, Ribeirao Preto, SP, Brazil, were used in this study. The viruses were propagated in C6/36 (Aedes albopictus mosquito cell line) cells and titrated by plaque assay in Vero E6 cells (an African green monkey kidney epithelium cell line) [4]. VERO E6 and C6/36 cells were maintained in Leibovitz’s medium (L-15) with 10% fetal bovine serum (FBS) at 37 °C and 28 °C, respectively. The viral titers were expressed as plaque-forming units per milliliter (PFU/ml).
Expression of the PLA2-CB isoforms
The nucleotide sequences of the PLA2-CB isoform genes were obtained from the GenBank database (PLA2-CB1, GenBank accession no. X12603.1, and PLA2-CB2, GenBank accession no. X16100.1). Both of these sequences represent the gene encoding the subunit precursor protein, which includes a peptide signal of 16 amino acid residues followed by 122 amino acid residues corresponding to the mature PLA2-CB isoform [3]. In this study, we used the nucleotide sequences encoding the mature PLA2-CB isoforms. The PLA2-CB1 and PLA2-CB2 nucleotide sequences were optimized using the preferred codons for expression in a prokaryotic system and synthesized by Genscript (USA) (Supplemental Figures 1 and 2). The optimized PLA2-CB isoform genes were cloned into the pGS-21a expression vector via restriction enzyme digestion (NdeI and XhoI) and DNA ligation (Genscript, USA). This vector allows the expression of recombinant proteins (rPLA2-CB1 and rPLA2-CB2) with a polyhistidine tag (6xHis) at the C-terminus for purification. The plasmids containing the PLA2-CB isoform genes (pGS21a-CB1 and pGS21a-CB2) were used to transform competent Escherichia coli (BL21(DE3) strain). One colony of E. coli containing each desired plasmid was grown in 400 mL of Luria-Bertani (LB) medium (pH 7.0) supplemented with 50 µg of ampicillin per mL at 37 °C with agitation at 200 rpm until an OD600 of 0.5-0.7 was reached. Then, expression of the rPLA2-CB isoforms was induced by the addition of 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), followed by incubation at 37 °C for 4 h with agitation at 200 rpm. The cell cultures were centrifuged at 6000 × g for 5 min at 4 °C, and the pellets were stored at -20 °C prior to use. A 1-g aliquot of the cell pellets was thawed on ice for 15 min and resuspended in 3 mL of lysis buffer (50 mM Tris-Cl, 1 mM EDTA, and 100 mM NaCl, pH 8.0). Then, lysozyme (1 mg/mL), RNase A (10 µg/mL) and DNase I (5 µg/mL) were added to the suspension and incubated on ice for 30 min. The suspension was sonicated on ice with five 30-s bursts and a 30-s interval between bursts (Sonifier Cell Disruptor Model SLPe, Branson, USA). The suspension was centrifuged at 6000 × g for 5 min at 4 °C, and the pellet and supernatant were analyzed by 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
Preparation of polyclonal antibodies against native PLA2-CB
A group of six-week-old BALB/c female mice (n = 10) was used to prepare polyclonal antibodies against native PLA2-CB. The animals were obtained from the Central Animal Facility at the University of Sao Paulo, Ribeirao Preto, Sao Paulo State, Brazil. Each animal was inoculated intraperitoneally with a solution containing native PLA2-CB (25 µg of PLA2-CB dissolved in 100 µL of 150 mM sodium chloride and mixed with 100 µl of complete Freund’s adjuvant). The animals were re-inoculated three times with one-week intervals between inoculations, with the same amount of PLA2-CB but with complete Freund’s adjuvant replaced with incomplete Freund’s adjuvant. One week after the last inoculation, the animals were anesthetized (100 mg/kg of ketamine and 20 mg/kg of xylazine) for blood collection by the retro-orbital route. The animals were euthanized after this procedure. The blood collected from all animals was pooled and centrifuged at 4500 rpm for 10 min at 4 °C to obtain hyperimmune serum samples. The serum samples were incubated at 56 °C for 40 min for complement inactivation. The inactivated hyperimmune serum samples were stored at -30 °C prior to use.
Analysis of recombinant protein expression
Expression of the rPLA2-CB1 and rPLA2-CB2 proteins was analyzed using a Western blotting assay. Briefly, an aliquot of the pellet, supernatant or whole cell lysate was subjected to 15% SDS-PAGE. Then, the proteins were electrotransferred (100 V for 1 h) to a nitrocellulose membrane using transfer buffer containing 39 mM glycine, 48 mM Tris base, 0.037% SDS, and 20% methanol. The blotting membrane was stained with Ponceau S staining buffer (0.2% Ponceau, 3% trichloroacetic acid, and 3% sulfosalicylic acid) for visualization and confirmation of protein transfer. The membrane was washed with distilled water and treated with blocking buffer (5% nonfat dry milk in Tris-buffered saline, TBS) for 2 h with continuous stirring at room temperature. The membrane was washed three times with TBS-Tween for 5 min with continuous stirring at room temperature. The membrane was incubated with a mouse hyperimmune serum (1:1000 dilution in blocking buffer) with continuous stirring at room temperature for 1 h. The membrane was washed three times with TBS-Tween for 5 min per wash and then incubated for 1 h at room temperature with an alkaline-phosphatase-conjugated goat anti-mouse IgG (whole molecule) secondary antibody (1:1000 dilution in blocking buffer; Sigma-Aldrich, Germany). The membrane was washed three times for 5 min per wash with TBS-Tween. The alkaline phosphatase substrate (BCIP/NBT, Sigma-Aldrich, Germany) was added directly to the membrane and maintained for up to 5 min or until the desired color was observed.
Refolding and purification of the recombinant proteins
The rPLA2-CB1 and rPLA2-CB2 proteins were expressed as insoluble inclusion bodies in E. coli; therefore, they were purified under denaturing conditions using a 1-mL Ni-NTA Superflow Cartridge (QIAGEN, Germany) in an Äkta FPLC System (GE Healthcare, USA). The cell lysate pellet (0.25 g) was frozen at -20 °C overnight, thawed on ice for 15 min, and resuspended in 5 mL of solubilization buffer (8 M urea, 100 mM NaH2PO4, 100 mM Tris-Cl, 1 mM PMSF, and 10 mM DTT, pH 8.0). The suspension was stirred for 1 h at room temperature and then centrifuged at 6000 × g for 40 min at room temperature. The supernatant containing the solubilized proteins was applied to an Ni-NTA Superflow Cartridge at a flow rate of 0.5 mL/min for recombinant protein binding. To induce proper folding of the recombinant proteins, a refolding step was performed while the proteins were linked to the Ni-NTA resin as follows: A decreasing linear gradient of urea and DTT concentrations in combination with CHAPS detergent (6-0 M urea, 10-0 mM DTT, 500 mM NaCl, 20 mM Tris-Cl, and 0.5% CHAPS, pH 7.0) in a total volume of 120 ml was applied to the Ni-NTA Superflow Cartridge at a flow rate of 0.1 ml/min. Then, the proteins were eluted with an elution buffer containing 1.5 M imidazole, 100 mM NaH2PO4, 20 mM Tris-Cl, and 0.5% CHAPS (pH 7.0). Fractions of the eluted solution were collected at a rate of 1 ml/min. After elution, the column was washed with 0.5 M NaOH to release non-eluted proteins. All fractions were analyzed using 15% SDS-PAGE. The fractions containing the recombinant proteins were pooled and subjected to a buffer exchange step (500 mM NaCl, 20 mM Tris-Cl, and 0.5% CHAPS, pH 7.0) to remove imidazole using a 15-ml Vivaspin 3-kDa MWCO concentrator (Sartorius, Germany), which was centrifuged at 3900 × g for 40 min at 24 °C. The filtrate was discarded, and the final recombinant protein solution was concentrated to a final volume of ~300 µl. The concentration of recombinant PLA2-CB was determined using a Bio-Rad Protein Assay Kit II (Bio-Rad, EUA), following the manufacturer’s recommendations. The proteins were stored at 4 °C prior to use.
Cytotoxicity of the recombinant proteins
The cytotoxicity of the rPLA2-CB1 and rPLA2-CB2 proteins was evaluated in Vero E6 cells using an MTT ([3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide]; Sigma–Aldrich, Germany) colorimetric assay [26]. Viable cells with active metabolism convert MTT into a purple-colored formazan product with an absorbance maximum near 570 nm. When the cells die, they lose the ability to convert MTT into formazan, and thus color formation serves as a useful and convenient marker for identifying viable cells. Confluent Vero E6 cell monolayers in 96-well plates were exposed to serial twofold dilutions of rPLA2-CB1, rPLA2-CB2, a pool of the recombinant proteins (50-0.4 µg/µL), and the buffer used in the buffer exchange step (CHAPS buffer). The cells were incubated for seven days at 37 °C. Then, 50 µL of L-15 containing MTT (final concentration, 1 mg/mL) was added to each well. After 4 h of incubation at 37 °C, the supernatant was removed, and 100 µL of dimethyl sulfoxide (DMSO) was added to each well to solubilize the formazan crystals. After shaking, the absorbance was measured at 540 nm. The concentration of each compound that reduced cell viability by 50% (CC50) was calculated by comparison of the absorbance of the treated cells with the absorbance of the untreated cells.
Virucidal activity of the recombinant proteins
The virucidal activity of the recombinant proteins against CHIKV, DENV-2, YFV and ZIKV was assessed as described previously [27]. Briefly, 10,000 PFU of each virus was incubated with serial twofold dilutions of rPLA2-CB1 (initial concentration, 100 µg/µl), rPLA2-CB2 (initial concentration, 100 µg/µl) for 1 hour at 37 °C. Viruses treated with native PLA2-CB (0.8 ng/µl) and CHAPS (0.5%) and untreated viruses were used as the controls. The mixtures were diluted 100 times to inoculate the Vero E6 cells with non-cytotoxic concentrations of the proteins and 100 PFU of each virus. After 1 h of incubation at 37 °C, the supernatant was removed, and the cell monolayers were overlaid with 1 ml of L-15 medium containing 2% FBS and 1.8% carboxymethylcellulose. After 7 days of incubation at 37 °C, the semisolid overlay medium was removed, and the cells were fixed and stained with naphthol blue-black in 5% acetic acid to count the plaques indicating cell lysis. Each experiment was repeated three times. The number of plaques observed in the cells infected with the treated viruses was compared with the number of plaques found in the cells infected with the untreated viruses to determine the percentage of plaque reduction. The 50% effective concentration (EC50), which is the concentration of samples that reduces the number of plaques by 50%, was determined in each antiviral assay by nonlinear regression analysis. Moreover, the selectivity index (SI, CC50/IC50) was calculated for each recombinant protein against each virus.
Phospholipase activity of the recombinant proteins
A colorimetric assay for PLA2 activity was performed as described previously [18, 25]. Briefly, PLA2 substrate micelles were prepared by sonication (Sonifier Cell Disruptor, model SLPe, Branson, EUA; 70% amplitude for 2 min) of a solution containing phosphatidylcholine as the phospholipase substrate and phenol red as a pH indicator (55 µM phenol red, 4 mM sodium cholate, 3.5 mM phosphatidylcholine, 100 mM NaCl, and 10 mM CaCl2 at pH 7.6). A 0.5-µg aliquot of PLA2-CB, rPLA2-CB1, or rPLA2-CB2 recombinant protein was added to a cuvette containing 1 ml of micelles at room temperature. The absorbance of the solution at 558 nm was measured at 5-min intervals for 15 minutes, using a Visible Spectrophotometer Model 722G, Global Trade Technology). The phospholipase activity of the recombinant proteins was confirmed when a reduction in absorbance was observed.
Results
Expression of the PLA2-CB isoforms in a prokaryotic system
DNA containing the optimized nucleotide sequences of the PLA2-CB1 and PLA2-CB2 genes was chemically synthesized and inserted into a plasmid vector (pGS-21a) that allowed expression of the encoded recombinant protein with a polyhistidine (6xHis) tag at the C-terminus for protein purification. These plasmid constructs (pPLA2-CB1 and pPLA2-CB2) were used to transform chemically competent E. coli BL21(DE3) cells for expression of the rPLA2-CB1 and rPLA2-CB2 proteins. SDS-PAGE analysis of each E. coli cell lysate showed the induction of expression of a ~16-kDa protein, which corresponds to the expected molecular mass of both the rPLA2-CB1 and rPLA2-CB2 proteins (Fig. 1). The E. coli cell lysates were analyzed by Western blotting, using a mouse hyperimmune serum that was raised against the native PLA2-CB as the primary antibody. The polyclonal antibodies in the hyperimmune serum were able to detect both of the induced proteins with a molecular mass of ~16 kDa, thereby confirming expression of both the rPLA2-CB1 and rPLA2-CB2 proteins (Fig. 2).

Analysis of rPLA2-CB1 and rPLA2-CB2 protein expression in E. coli strain BL21(DE3). The bacteria were transformed with the pPLA2-CB1 and pPLA2-CB2 plasmids, and rPLA2-CB1 (A) and rPLA2-CB2 (B) protein expression was analyzed by 15% SDS-PAGE. Proteins with a molecular mass of ~16 kDa (red arrows) were observed after 2 (lanes 2 and 7) and 4 (lanes 4 and 8) hours of IPTG induction. The bacteria transformed with pPLA2-CB1 and pPLA2-CB2 but without IPTG induction are shown in lanes 1-2 and 5-6, respectively. Lane M shows the molecular mass markers (BenchMark Protein Ladder, Invitrogen, USA). The red arrows indicate the recombinant proteins

Identification of the rPLA2-CB1 and rPLA2-CB2 proteins. The proteins from E. coli BL21(DE3) cells transformed with the pPLA2-CB1 and pPLA2-CB2 plasmids were analyzed in a Western blot assay using mouse polyclonal antibodies against the native PLA2-CB. The whole lysates from E. coli transformed with pPLA2-CB1 and pPLA2-CB2 but without IPTG induction are shown in lanes 1 and 2, respectively. The whole lysates from E. coli transformed with pPLA2-CB1 and pPLA2-CB2 after 2 hours of induction with IPTG are shown in lanes 4 and 5, respectively. Lane 3 contains native PLA2-CB. Lane M shows the molecular mass markers (PageRuler Prestained Protein Ladder, Thermo Fisher Scientific, USA). The red arrows indicate the recombinant proteins. The dotted red arrows indicate the oligomers of the recombinant proteins. The yellow arrow indicates the native PLA2-CB
To examine the solubility of the recombinant proteins, the E. coli lysate supernatants and pellets were analyzed by SDS-PAGE (Fig. 3). The rPLA2-CB1 and rPLA2-CB2 proteins were found in the pellets of the cell lysates, indicating they were expressed as insoluble proteins.

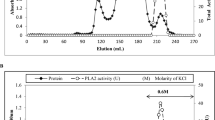
Refolding and purification of the rPLA2-CB1 protein. (A) Chromatographic profile of the rPLA2-CB1 protein refolding and purification steps. The blue line shows the absorbance (Mau) of the collected fractions at 280 nm. The green line shows the decreasing linear gradients of the urea (6-0 M) and DTT (10-0 mM) concentrations applied to the column. The x-axis shows the numbers of the fractions collected (red) and the volume (mL) (black) of the mobile phase applied to the column. (B) Zoom of the elution step and column washing with 0.5 M NaOH. (C) 15% SDS-PAGE of the collected fractions. Lane M, BenchMark Protein Ladder (Invitrogen, USA); lane 1, solubilized E. coli lysate pellet applied to the column; lane 2, fraction 1, unbound proteins; lane 3, elution fraction 16; lane 4, elution fraction 17; lane 5, elution fraction 18; lane 6, elution fraction 19; lane 7, elution fraction 20; lane 8, elution fraction 21; lane 9, washed (0.5 M NaOH) fraction 24. Red arrows indicate the eluted rPLA2-CB1 protein
Refolding and purification of the recombinant PLA2-CB isoforms
The insoluble rPLA2-CB1 and rPLA2-CB2 proteins were purified under denaturing conditions. The proteins contained in the pellets of the E. coli lysates were solubilized with urea and applied to an Ni-NTA column for binding of the rPLA2-CB1 and rPLA2-CB2 proteins. A decreasing linear gradient of urea and DTT concentrations containing CHAPS detergent as a protein stabilizer was applied to the column while the proteins were bound to the Ni-NTA resin to allow refolding. Then, the rPLA2-CB1 and rPLA2-CB2 proteins were eluted with a solution containing imidazole and CHAPS. Proteins with a molecular mass of ~16 kDa, corresponding to the expected molecular mass of both the rPLA2-CB1 and rPLA2-CB2 proteins, were obtained (Fig. 3 and 4). We tried to remove the CHAPS detergent from the eluted solution by dialysis, but this resulted in protein precipitation (data not shown). Therefore, we decided to use a solution containing the recombinant proteins and CHAPS in the virucidal assay. The protein concentrations were determined after purification and found to be 10 and 50 μg/μl for the rPLA2-CB1 and rPLA2-CB2 protein, respectively.

Refolding and purification of the rPLA2-CB2 protein. (A) Chromatographic profile of the rPLA2-CB2 protein during the refolding and purification steps. The blue line shows the absorbance (mAu) of the fractions at 280 nm. The green line shows the decreasing linear gradient of the urea (6-0 M) and DTT (10-0 mM) concentrations applied to the column. The x-axis shows the numbers of the fractions collected (red) and the volume (mL) (black) of the mobile phase applied to the column. (B) Zoom of the elution step and column washing with 0.5 M NaOH. (C) 15% SDS-PAGE of the collected fractions. Lane M, BenchMark Protein Ladder (Invitrogen, USA); lane 1, solubilized E. coli lysate pellet applied to the column; lane 2, elution fraction 1; lane 3, elution fraction 2; lane 4, elution fraction 15; lane 5, elution fraction 16; lane 6, eluted fraction 17; lane 7, eluted fraction 18; lane 8, eluted fraction 19; lane 9, washed (0.5 M NaOH) fraction 24. Red arrows show the eluted rPLA2-CB2 protein
Cytotoxicity of the recombinant PLA2-CB isoforms
The cytotoxicity of the rPLA2-CB1 and rPLA2-CB2 proteins was evaluated in Vero E6 cells using an MTT colorimetric assay. The cytotoxicity of CHAPS, included in the elution solution, was also examined. The cells were treated with twofold serial dilutions of each recombinant protein or CHAPS for seven days, and the CC50 values were calculated (Table 1).
Virucidal activity of the recombinant proteins
We previously showed that PLA2-CB isolated from Crotalus durissus terrificus venom exerted inhibitory effects by direct action on the viral envelope [28]; therefore, we analyzed the inhibitory activity of the recombinant proteins against CHIKV, DENV-2, YFV and ZIKV in a virucidal assay. Both the rPLA2-CB1 and rPLA2-CB2 proteins showed a dose-dependent virucidal effect against all tested viruses (Table 2). Complete inhibition of Vero cell infection was observed when the highest recombinant protein concentrations and 0.8 ng/µL of native PLA2-CB were used. Conversely, no inhibition was observed when the viruses were treated with 0.5% CHAPS. The mean inhibition of plaque formation (Table 2) induced by the proteins was used to calculate the EC50 by nonlinear regression analysis (Table 3, Online Resource 1). Both recombinant proteins yielded low EC50 values against each virus. Moreover, the recombinant proteins showed high SI values for each virus (Table 3).
Phospholipase activity of the recombinant proteins
Previous studies have suggested that the enzymatic activity of native PLA2-CB is important for its antiviral effect [28, 35]. Therefore, we examined the phospholipase activity of the rPLA2-CB1 and rPLA2-CB2 proteins, using a colorimetric assay with phosphatidylcholine as the phospholipase target. Hydrolysis of phospholipids at the sn-2 position generates lysophospholipids and free fatty acids, which decrease the pH of the solution. Therefore, the hydrolysis of phosphatidylcholine induced by the rPLA2-CB1 and rPLA2-CB2 proteins was measured by monitoring the change in absorbance at 558 nm for up to 15 minutes (Fig. 5). Decreasing absorbance over this period was observed when the phosphatidylcholine was treated with each of the recombinant proteins, confirming their phospholipase activity.

Phospholipase activity of the rPLA2-CB1 and rPLA2-CB2 proteins. Phosphatidylcholine hydrolysis was monitored by measuring the absorbance of the solution at 580 nm at 5-min intervals for up to 15 min following treatment with 1 μg of each of the rPLA2-CB1, rPLA2-CB2, and native PLA2-CB proteins
Discussion
We previously conducted a proof-of-concept study to demonstrate the antiviral activity of the PLA2-CB isolated from Crotalus durissus terrificus venom [27]. However, this highly toxic compound would be very unlikely to be used in the treatment of virus infections. Therefore, we aimed to produce a PLA2-CB in vitro that maintains its viral inhibitory activity. This PLA2-CB could become a promising lead compound that can be subjected to amino acid sequence modification to produce less-toxic compounds that retain their antiviral activity. In this study, we describe the expression in a prokaryotic system and the virucidal activity of two recombinant isoforms of the PLA2-CB from Crotalus durissus terrificus venom. E. coli is the most frequently used protein expression host due to its extensive genetic characterization, low cost, easy handling, and high protein yield. In this study, the E. coli BL21(DE3) strain was selected for the expression of the rPLA2-CB1 and rPLA2-CB2 proteins (Fig. 2). The detection of proteins with a higher molecular mass in a Western blot assay suggested the presence of oligomers of the rPLA2-CB1 and rPLA2-CB2 proteins (~32 kDa), which probably appeared due to the high expression level of the proteins in E. coli. The BL21(DE3) strain uses the bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes [44]. Many extracellular proteins, such as sPLA2s, stabilize their structural conformation through disulfide bonds, which are formed in an oxidative reaction between the thiol groups (-SH) of two cysteine residues. The cytoplasm of prokaryotic and eukaryotic cells has a reducing environment, which prevents the formation of disulfide bonds. Disulfide bond formation in eukaryotic cells occurs in the lumen of the endoplasmic reticulum, which has a more oxidizing environment [27]. Prokaryotic cells do not have a subcellular compartment that provides the oxidizing environment required for the proper formation of these disulfide bonds; thus, recombinant proteins expressed in this system tend to accumulate in inclusion bodies due to exposure of hydrophobic sites induced by misfolding, resulting in the production of aggregates of insoluble proteins [6, 12]. Both PLA2-CB isoforms have seven disulfide bonds that play a key role in stabilizing the tertiary structure of these proteins [13]. In this study, rPLA2-CB1 and rPLA2-CB2 were expressed as insoluble proteins, as demonstrated by the formation of aggregates induced by misfolding. Soluble recombinant proteins can be obtained in prokaryotic systems by optimization of expression variables, such as the temperature, IPTG concentration, and time of expression induction [43]. We tried to optimize these variables, but insoluble proteins were always obtained (data not shown). Other recombinant PLA2 proteins have been expressed in prokaryotic systems as insoluble proteins, which were purified under denaturing conditions, followed by dialysis or gel filtration for refolding [7, 8, 39, 41, 46]. During protein refolding, insoluble molecules can be formed as a result of inappropriate intra- and inter-molecular rearrangements. These non-native interactions occur due to exposure of hydrophobic sites and/or intramolecular disulfide bonds [15]. The seven disulfide bonds of PLA2-CB, in addition to numerous hydrophobic amino acid residues (41 and 40 in the PLA2-CB1 and PLA2-CB2 isoform, respectively) [14] can contribute to protein aggregation during the refolding step when using dialysis or gel filtration. Generally, these refolding strategies are very rapid and difficult to control, which contributes to the formation of non-native molecular interactions and, consequently, protein aggregation and precipitation [15, 21]. In the present study, we used a novel single-step strategy for refolding and purification of the insoluble rPLA2-CB1 and rPLA2-CB2 proteins. The solubilized proteins were first linked to a nickel-containing column; then, a decreasing linear gradient of urea and DTT concentrations at an optimized flow rate was applied to the column to induce correct folding of the proteins, after which the refolded recombinant proteins were eluted. We used the detergent CHAPS as a protein stabilizer during the refolding and elution steps to avoid protein aggregation. CHAPS is a zwitterionic surfactant non-denaturing detergent that is capable of preventing protein aggregation and preserving the enzymatic activity of proteins in solution [1, 20]. We tried to remove CHAPS from the eluates containing the recombinant proteins by dialysis, but this resulted in precipitation of the proteins. We therefore, decided to use the eluate containing the rPLA2-CB1 and rPLA2-CB2 proteins in the presence of CHAPS in the virus inhibition assay. We previously assessed the viral inhibitory effect of the PLA2-CB isolated from Crotalus durissus terrificus venom using several methodological strategies and found the highest inhibitory effect in the virucidal assay [27]. We found evidence that the phospholipase activity of PLA2-CB might be responsible for the cleavage of glycerophospholipids at the lipid bilayer envelope, resulting in virus inactivation [28, 35]. Therefore, in the present study, the recombinant isoforms of PLA2-CB were evaluated only in a virucidal assay to evaluate the inhibitory effect of the proteins against CHIKV, DENV-2, YFV, and ZIKV. A control virucidal experiment showed no inhibitory effect of CHAPS, which confirmed that the virucidal activity was caused by the recombinant proteins. The levels of virucidal activity for both the rPLA2-CB1 and rPLA2-CB2 proteins were lower than those observed previously with the native PLA2-CB (EC50, 240-260 ng/µL for the recombinant proteins and 0.01647-0.044 for the native PLA2-CB) [27]. Previous studies have shown that amino acid sequence differences between the PLA2-CB1 and PLA2-CB2 isoforms result in differences in enzymatic and pharmacological activity [12, 14]. The amino acid sequences of the rPLA2-CB1 and rPLA2-CB2 proteins differ somewhat from those of the native proteins. The optimized nucleotide sequences of the PLA2-CB1 and PLA2-CB2 genes were cloned into the expression vector pG21 flanked by recognition sequences for the restriction enzymes NdeI and XhoI. The NdeI restriction site (5’CATATG3’) added a start codon for the expression of the PLA2-CB1 and PLA2-CB2 genes, resulting in the inclusion of a methionine (M) residue at the N-terminal end of the proteins, whereas the XhoI restriction site (5’CTCGAG3’) resulted in the addition of leucine (L) and glutamic acid (E) amino acid residues at the C-terminal end of the proteins. Finally, the rPLA2-CB1 and rPLA2-CB2 proteins were expressed with a 6-His tag at the C-terminal end (Supplementary Figure 3). The lower phospholipase activity of the recombinant proteins when compared to the native PLA2-CB may be due to the nine additional amino acid residues present in their sequences, which in turn may have resulted in lower virucidal activity. The recombinant proteins also showed higher cytotoxicity in Vero E6 cells than the native PLA2-CB protein (CC50, 10.03-14.74 µg/µL for the recombinant proteins and <0.5 µg/µL for the native PLA2-CB) [27]. The presence of the detergent CHAPS in the elution solution may be responsible for the higher cytotoxicity found for the recombinant proteins. Further studies are needed to determine how to obtain soluble rPLA2-CB1 and rPLA2-CB2 proteins without CHAPS to reduce the cytotoxicity of the recombinant proteins.
In summary, we expressed the rPLA2-CB1 and rPLA2-CB2 proteins in a prokaryotic system and purified them using an innovative in-column refolding method. The recombinant proteins showed virucidal effects against CHIKV, DENV-2, YFV and ZIKV, suggesting that they could be used as a model for the development of broad-spectrum antiviral compounds.
Abbreviations
- DENV:
-
Dengue virus
- ZIKV:
-
Zika virus
- YFV:
-
Yellow fever virus
- CHIKV:
-
Chikungunya virus
- PLA2 :
-
Phospholipase A2
- sPLA2 :
-
Secreted phospholipase A2
- svPLA2 :
-
Snake venom phospholipase A2
- CA:
-
Crotapotin
- PLA2-CB:
-
Phospholipase A2 crotoxin B
- PLA2-CB1:
-
Phospholipase A2 crotoxin B isoform 1
- PLA2-CB2:
-
Phospholipase A2 crotoxin B isoform 2
- rPLA2-CB1:
-
Recombinant phospholipase A2 crotoxin B isoform 1
- rPLA2-CB2:
-
Phospholipase A2 crotoxin B isoform 2
- sn-2:
-
Stereo-specific number 2
- 6xHis:
-
Polyhistidine tag
- LB:
-
Luria-Bertani medium
- TBS:
-
Tris-buffered saline
- MTT:
-
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- DMSO:
-
Dimethyl sulfoxide
- CC50 :
-
50% cytotoxic concentration
- PFU:
-
Plaque-forming units
- IPTG:
-
Isopropyl β-D-1-thiogalactopyranoside
- BCIP/NBT:
-
5-Bromo-4-chloro-3-indolyl-phosphate/nitro blue tetrazolium
- PMSF:
-
Phenylmethylsulfonyl fluoride
- DTT:
-
Dithiothreitol
- SDS:
-
Sodium dodecyl sulfate
- PAGE:
-
Polyacrylamide gel electrophoresis
- IgG:
-
Immunoglobulin G
- SD:
-
Standard deviation
- CHAPS:
-
3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate
References
Alibolandi M, Mirzahoseini H (2011) Chemical assistance in refolding of bacterial inclusion bodies. Biochem Res Int 2011:631607
Amaral JK, Schoen RT (2018) Chikungunya in Brazil: rheumatologists on the front line. J Rheumatol 45:1491–1492
Bouchier C, Boulain JC, Bon C, Ménez A (1991) Analysis of cDNAs encoding the two subunits of crotoxin, a phospholipase A2 neurotoxin from rattlesnake venom: the acidic non enzymatic subunit derives from a phospholipase A2-like precursor. Biochim Biophys Acta 1088:401–408
Burleson FG, Chambers TM, Wiedbrauk DL (1992) Virology: a laboratory manual. Academic Press, San Diego
Castillo JC, Vargas LJ, Segura C, Gutiérrez JM, Pérez JC (2012) In vitro antiplasmodial activity of phospholipases A2 and a phospholipase homologue isolated from the venom of the snake Bothrops asper. Toxins (Basel) 4:1500–1516
Chen YH, Wang YM, Hseu MJ, Tsai IH (2004) Molecular evolution and structure-function relationships of crotoxin-like and asparagine-6-containing phospholipases A2 in pit viper venoms. Biochem J 381:25–34
Chioato L, Ward RJ (2003) Mapping structural determinants of biological activities in snake venom phospholipases A2 by sequence analysis and site directed mutagenesis. Toxicon 42:869–883
Chiou YL, Cheng YC, Kao PH, Wang JJ, Chang LS (2008) Mutations on the N-terminal region abolish differentially the enzymatic activity, membrane-damaging activity and cytotoxicity of Taiwan cobra phospholipase A2. Toxicon 51:270–279
Collins ND, Barrett AD (2017) Live attenuated yellow fever 17D vaccine: a legacy vaccine still controlling outbreaks in modern day. Curr Infect Dis Rep 19:14
Dennis EA, Cao J, Hsu YH, Magrioti V, Kokotos G (2011) Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem Rev 111:6130–6185
Doley R, Kini RM (2009) Protein complexes in snake venom. Cell Mol Life Sci 66:2851–2871
Faure G, Harvey AL, Thomson E, Saliou B, Radvanyi F, Bon C (1993) Comparison of crotoxin isoforms reveals that stability of the complex plays a major role in its pharmacological action. Eur J Biochem 214:491–496
Faure G, Choumet V, Bouchier C, Camoin L, Guillaume JL, Monegier B, Vuilhorgne M, Bon C (1994) The origin of the diversity of crotoxin isoforms in the venom of Crotalus durissus terrificus. Eur J Biochem 223:161–164
Faure G, Xu H, Saul FA (2011) Crystal structure of crotoxin reveals key residues involved in the stability and toxicity of this potent heterodimeric β-neurotoxin. J Mol Biol 412:176–191
Ferré H, Ruffet E, Nielsen LL, Nissen MH, Hobley TJ, Thomas OR, Buus S (2005) A novel system for continuous protein refolding and on-line capture by expanded bed adsorption. Protein Sci 14:2141–2153
Figueiredo LT (2015) The recent arbovirus disease epidemic in Brazil. Rev Soc Bras Med Trop. https://doi.org/10.1590/0037-8682-0179-2015
Figueiredo LT (2016) How are so many foreign arboviruses introduced in Brazil? Rev Soc Bras Med Trop. https://doi.org/10.1590/0037-8682-0499-2016
Francischetti IM, Gombarovits ME, Valenzuela JG, Carlini CR, Guimarães JA (2000) Intraspecific variation in the venoms of the South American rattlesnake (Crotalus durissus terrificus). Comp Biochem Physiol C Toxicol Pharmacol 127:23–36
Hendon RA, Fraenkel-Conrat H (1971) Biological roles of the two components of crotoxin. Proc Natl Acad Sci USA 68:1560–1563
Hjelmeland LM (1980) A nondenaturing zwitterionic detergent for membrane biochemistry: design and synthesis. Proc Natl Acad Sci USA 77:6368–6370
Jaenicke R (1982) Folding and association of proteins. Biophys Struct Mech 8:231–256
Kang TS, Georgieva D, Genov N, Murakami MT, Sinha M, Kumar RP, Kaur P, Kumar S, Dey S, Sharma S, Vrielink A, Betzel C, Takeda S, Arni RK, Singh TP, Kini RM (2011) Enzymatic toxins from snake venom: structural characterization and mechanism of catalysis. FEBS J 278:4544–4576
Long KC, Ziegler SA, Thangamani S, Hausser NL, Kochel TJ, Higgs S, Tesh RB (2011) Experimental transmission of Mayaro virus by Aedes aegypti. Am J Trop Med Hyg 85:750–757
Lowe R, Barcellos C, Brasil P, Cruz OG, Honório NA, Kuper H, Carvalho MS (2018) The Zika virus epidemic in Brazil: from discovery to future implications. Int J Environ Res Public Health 15(1):96
Lôbo de Araújo A, Radvanyi F (1987) Determination of phospholipase A2 activity by a colorimetric assay using a pH indicator. Toxicon 25:1181–1188
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63
Muller VD, Russo RR, Cintra AC, Sartim MA, ReM Alves-Paiva, Figueiredo LT, Sampaio SV, Aquino VH (2012) Crotoxin and phospholipases A2 from Crotalus durissus terrificus showed antiviral activity against dengue and yellow fever viruses. Toxicon 59:507–515
Muller VD, Soares RO, dos Santos NN, Trabuco AC, Cintra AC, Figueiredo LT, Caliri A, Sampaio SV, Aquino VH (2014) Phospholipase A2 isolated from the venom of Crotalus durissus terrificus inactivates dengue virus and other enveloped viruses by disrupting the viral envelope. PLoS One 9:e112351
Murakami M, Sato H, Miki Y, Yamamoto K, Taketomi Y (2015) A new era of secreted phospholipase A2 (sPLA2). J Lipid Res 56:1248–1261
Nunes DC, Figueira MM, Lopes DS, De Souza DL, Izidoro LF, Ferro EA, Souza MA, Rodrigues RS, Rodrigues VM, Yoneyama KA (2013) BnSP-7 toxin, a basic phospholipase A2 from Bothrops pauloensis snake venom, interferes with proliferation, ultrastructure and infectivity of Leishmania (Leishmania) amazonensis. Parasitology 140:844–854
Pereañez JA, Gómez ID, Patiño AC (2012) Relationship between the structure and the enzymatic activity of crotoxin complex and its phospholipase A2 subunit: an in silico approach. J Mol Graph Model 35:36–42
Quach ND, Arnold RD, Cummings BS (2014) Secretory phospholipase A2 enzymes as pharmacological targets for treatment of disease. Biochem Pharmacol 90:338–348
Reeks TA, Fry BG, Alewood PF (2015) Privileged frameworks from snake venom. Cell Mol Life Sci 72:1939–1958
Rodrigues RS, Izidoro LF, de Oliveira RJ, Sampaio SV, Soares AM, Rodrigues VM (2009) Snake venom phospholipases A2: a new class of antitumor agents. Protein Pept Lett 16:894–898
Russo RR, Müller VDM, Cintra ACO, Figueiredo LTM, Sampaio SV, Aquino VH (2014) Phospholipase A2 crotoxin B isolated from the venom of Crotalus durissus terrificus exert antiviral effect against dengue virus and yellow fever virus through its catalytic activity. J Virol Antivir Res 3:1
Rübsamen K, Breithaupt H, Habermann E (1971) Biochemistry and pharmacology of the crotoxin complex. I. Subfractionation and recombination of the crotoxin complex. Naunyn Schmiedebergs Arch Pharmacol 270:274–288
Sakkas H, Bozidis P, Franks A, Papadopoulou C (2018) Oropouche fever: a review. Viruses 10(4):175
Samy RP, Gopalakrishnakone P, Stiles BG, Girish KS, Swamy SN, Hemshekhar M, Tan KS, Rowan EG, Sethi G, Chow VT (2012) Snake venom phospholipases A(2): a novel tool against bacterial diseases. Curr Med Chem 19:6150–6162
Seto M, Ogawa T, Kodama K, Muramoto K, Kanayama Y, Sakai Y, Chijiwa T, Ohno M (2008) A novel recombinant system for functional expression of myonecrotic snake phospholipase A(2) in Escherichia coli using a new fusion affinity tag. Protein Expr Purif 58:194–202
Sharp TM, Tomashek KM, Read JS, Margolis HS, Waterman SH (2017) A new look at an old disease: recent insights into the global epidemiology of dengue. Curr Epidemiol Rep 4:11–21
Silveira LB, Marchi-Salvador DP, Santos-Filho NA, Silva FP, Marcussi S, Fuly AL, Nomizo A, da Silva SL, Stábeli RG, Arantes EC, Soares AM (2013) Isolation and expression of a hypotensive and anti-platelet acidic phospholipase A2 from Bothrops moojeni snake venom. J Pharm Biomed Anal 73:35–43
Slotta KH, Fraenkel-Conrat H (1938) Schlangengiffe, III: mitteilung reiningung und crystallization des klappershclangengiffes. Berichte der Deutschen Chemischen Gesellschaft 71:1076–1081
Smith HE (2007) The transcriptional response of Escherichia coli to recombinant protein insolubility. J Struct Funct Genom 8:27–35
Studier FW, Moffatt BA (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol 189:113–130
Wilder-Smith A, Gubler DJ, Weaver SC, Monath TP, Heymann DL, Scott TW (2017) Epidemic arboviral diseases: priorities for research and public health. Lancet Infect Dis 17:e101–e106
Yunes Quartino PJ, Barra JL, Fidelio GD (2012) Cloning and functional expression of secreted phospholipases A(2) from Bothrops diporus (Yarará Chica). Biochem Biophys Res Commun 427:321–325
Funding
This work was supported by the Sao Paulo Research Foundation (FAPESP), grant no. 2014/02438-6) and the National Council of Technological and Scientific Development (CNPq) (grant no. 479512/2012-4). RR was supported by a FAPESP scholarship (grant no. 2012/12605-1) and VHA holds a CNPq-PQ fellowship (grant no. 306471/2017-5).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Animal experiments
All animal experiments were performed according to the guidelines of the Brazilian College of Animal Experimentation and were approved by the Ethical Committee on Animal Experimentation of the Campus of Ribeirao Preto, University of Sao Paulo (CEUA/USP-RP, Permit. Nº 12.1.1854.53.8).
Additional information
Handling Editor: Patricia Aguilar.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Linear regression plots for inhibition of plaque formation (percent) in Vero E6 cells infected with YFV, CHIKV, DENV-2 or ZIKV, which were treated with various concentrations of rPLA2-CB1 and rPLA2-CB2 proteins
Rights and permissions
About this article

Cite this article
Russo, R.R., dos Santos Júnior, N.N., Cintra, A.C.O. et al. Expression, purification and virucidal activity of two recombinant isoforms of phospholipase A2 from Crotalus durissus terrificus venom. Arch Virol 164, 1159–1171 (2019). https://doi.org/10.1007/s00705-019-04172-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-019-04172-6