
Abstract
We report on a lateral flow immunoassay (LFIA) for influenza A antigen using fluorescently-doped silica nanoparticles as reporters. The method is taking advantage of the high brightness and photostability of silica nanoparticles (doped with the dye Cy5) and the simplicity and rapidity of LFIA. The nucleoprotein of influenza A virion (one of its most abundant structural proteins) was used as a model to demonstrate a performance of the LFIA. Under optimized conditions and by using a portable strip reader, the fluorescence-based LFIA is capable of detecting a recombinant nucleoprotein as low as 250 ng · mL-1 using a sample volume of 100 μL, within 30 min, and without interference by other proteins. The successful detection of the nucleoprotein in infected allantoic fluid demonstrated the functionality of the method. By comparison with a commercial influenza A test based on gold nanoparticles as reporters, the system provides an 8-fold better sensitivity.

A rapid and sensitive lateral flow immunoassay for influenza A antigen was developed using fluorescently-doped silica nanoparticles. A sample containing nucleoprotein as a target analyte induced an accumulation of the fluorescent conjugates at the test spot. The signal was then measured quantitatively using a portable strip reader.
Similar content being viewed by others

Introduction
Rapid and sensitive point-of-care (POC) detection remains a challenge in diagnostics and other biomedical applications. Various molecular diagnostic approaches and technologies have been developed to detect target analytes. These include enzyme-linked immunosorbent assay (ELISA) [1, 2], microarrays [3, 4], surface plasmon resonance (SPR) [5], surface enhanced Raman scattering (SERs) [6, 7], etc. Although these techniques offer a good sensitivity and selectivity, they often require sophisticated and high-cost instrumentation, laborious and time-consuming procedures, and trained operating personnel [8]. Recently, emerging lateral flow immunoassay (LFIA), also called a dry-reagent strip biosensor, has received increased interest as a tool for POC screening. The system offers a low-cost, one-step analysis as well as an easy-to-use platform, which can be applied for routine use in research and clinical applications [9–11].
Within the system, gold nanoparticles are commonly used as labels for visual detection, which is either qualitative or semiquantitative analysis [12]. However, the conventional systems based on gold nanoparticles as reporters have limitation in detecting the target analyte at low concentration [13]. Different systems using various labels have been developed in order to respond to this unmet need. These, for example, are dye-encapsulated immunoliposomes [14], carbon nanoparticles [15], and organic fluorophores [16]. Due to its intrinsic sensitivity, the fluorescence-based LFIA is a promising technique for improving sensitivity and capability in quantitative discrimination with the use of fluorophores [17].
Cy5 is proven to be one of the most important dyes that have been used as labeling in biological research fields such as fluorescence imaging, fluorescence biosensors, flow cytometry, microarray analysis, etc. [18]. Since the absorption and emission wavelength of Cy5 dyes are in the red spectral region, this allows an exclusion of the intrinsic fluorescence of most biological samples and the scatter of the system reacting, resulting in a lower background noise. However, it was reported that Cy5 dye suffered from significant photobleaching, and biodegradation under ozone and laser exposure as well as proton concentration [19–21], which limits its use in further applications. Recently, fluorescence-doped silica nanoparticles have shown advantages over organic fluorophores as highly sensitive and photostable fluorescence probes for bioanalysis [22]. During the synthesis, thousands of fluorescence molecules are trapped inside silica matrix protecting them from the oxygenic environment. As a result, dye photostability and probe sensitivity are greatly improved. By using conventional silica-based chemistry, specific functional groups could be easily added on particle surface to provide reactive sites for covalent bioconjugation which can prevent desorption of biomolecules from particle surface and control number as well as orientation of those molecules [23, 24]. Based on their unique optical properties, as well as, versatility in synthesis and surface modification, fluorescence-doped silica nanoparticles are predicted to be good candidates as fluorescence probes for a LFIA system.
We described here a fluorescence-based LFIA using Cy5-doped silica nanoparticles as reporters. The nucleoprotein (NP), one of the most conserved and abundant proteins present in the influenza A virion, was used as a target analyte to evaluate the performance of the biosensor system. Moreover, it was hypothesized that the system could detect the NP in the presence of complex sample matrix such as the infected allantoic fluid. By combining the efficiency and specificity of an immunoassay with the distinct properties of fluorescence-doped silica nanoparticles, one-step and sensitive LFIA developed in this work could have a good functionality for the analysis of a target protein. The system could also be useful for developing POC screening in medical diagnostic applications and offers potential therapeutic relevant decision.
Experimental
Reagents
Tetramethylorthosilicate (TEOS), aminopropyltriethoxy silane (APTES), 1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC), N-Hydroxy succinimide esters (NHS), Bovine Serum Albumin (BSA), and Polyvinyl pyrrolidone (PVP) were obtained from Sigma-Aldrich (MO, USA, http://www.sigmaaldrich.com), Cy5 NHS ester was purchased from Lumiprobe (FL, USA, http://www.lumiprobe.com). Carboxyethylsilanetriol disodium salt, 25 % in water, and 3-trihydroxysilylpropyl methylphosphonate (THPMP) were purchased from Gulf Chemical (Singapore, http://www.gulfchem.com.sg/), Dulbecco’s Phosphate-Buffered Saline (DPBS) was obtained from Invitrogen (NY, USA, http://www.invitrogen.com). Tween 20 and nonfat dry milk were obtained from Bio-Rad (CA, USA, http://www.bio-rad.com).
Monoclonal antibody specific to influenza A nucleoprotein (MAb), LFIA test strips with secondary polyclonal anti-mouse IgG antibody (Ab) immobilized at the control line (C line), and commercial Flu A test were supplied from Innova Biotechnology (Bangkok, Thailand, http://www.innovabiotechnology.com).
Source of recombinant nucleoprotein (rNP) and preparation of virus infected allantoic fluid
rNP used in this study was generated and characterized as described previously [25]. Ten-day-old embryonated specific pathogen-free chicken eggs (Suwanvajokkasikit Animal Research and Development Institute, Kasetsart University, Nakhon Pathom, Thailand) were inoculated with influenza virus A/Thailand/104/2009 (H1N1), provided by Prof. Pilaipan Puthavathana (Mahidol University, Bangkok, Thailand) according to a protocol described previously [26]. The allantoic fluid infected with influenza A virus was then inactivated using binary ethylenimine inactivation method [27] and purified using a centrifugation in a sucrose gradient [28]. All viral manipulations were performed under appropriate biosafety level 2 plus laboratory conditions.
Fluorescence-doped silica nanoparticle synthesis
Cy5-doped silica nanoparticles were prepared by modified Stöber method [29] using following procedures: Prior to the synthesis, all glass reaction vials were washed with 1 M NaOH solution for 15 min and rinsed with deionized water and ethanol to prevent the seeding of particles on the glass surface. Cy5 NHS ester (2.75 μmol) was dissolved in 500 μL of anhydrous DMSO and APTES was added at a molar ratio of 2:1 APTES:dye. The APTES was allowed to react with the dye solution for 1 h in the dark with vigorous stirring. EtOH (16.75 mL) and 25 % NH4OH (1.2 mL) were mixed in the glass vial. Under continuous stirring, the prepared dye-APTES conjugate was added to the mixture and left to react for 1 h. Next, TEOS (710 μL) was added to the mixture, and the reaction was continued for 24 h in the dark with continuous stirring. After centrifugation at 12,000 rpm for 20 min, the particles were washed three times with EtOH and three times with deionized water to remove free dyes and unreacted reagents. The particles were then resuspended in deionized water. The size of nanoparticles was then determined using transmission electron microscopy (TEM, JEM-2010, JEOL, Japan).
Surface modification of Cy5-doped silica nanoparticles
To introduce carboxyl groups on the surface of particles, 10 mg of the particles were diluted to a total volume of 1 mL with 10 mM DPBS. Carboxylsilane (20 μL) and THPMP (20 μL) were then added to the solution with continuous stirring 24 h in the dark. The modified particles were washed three times and resuspended in DPBS to a concentration of 10 mg·mL−1. The zeta potential was measured on a Malvern zetasizer (UK) using the Smoluchowski model to determine changes of surface charge after surface modification.
Conjugation of antibodies on carboxyl-modified particles
The antibodies were immobilized on the surface of carboxyl-modified particles through carbodiimide chemistry as described in following procedures: 1 mg of the particles was diluted in DPBS with a total volume of 500 μL. EDC (0.2 mg) and NHS (0.2 mg) were added to the particle solution and allowed to react for 15 min with gentle mixing. The activated particles were washed one time to remove unreacted chemicals and reconstituted in 500 μL of DPBS. MAb (20 μg) was added into the activated particles and mixed gently for 3 h at 4 °C. The conjugates were washed twice with DPBS and resuspended in 500 μL of a blocking solution (1 % BSA in 40 mM Tris–HCl) for 30 min at 4 °C. The blocked conjugates were then washed three times with DPBS and resuspended in a storage buffer (0.1 % BSA and 0.1 % PVP in 10 mM DPBS) at a final concentration of 10 mg·mL−1.
Fabrication of fluorescence-based LFIA
LFIA test strip was composed of 4 components including sample pad, conjugate pad, a Hi-Flow Plus HF180 nitrocellulose membrane (Merck Millipore, MA, USA, http://www.merckmillipore.com), and absorbent pad. Within the membrane, 1 mg·mL−1 secondary polyclonal anti-mouse Ab was immobilized as a horizontal line at what was defined as the C line. Further, MAb was spotted at the position at what was defined as the T spot, approximately 5 mm upstream from the C line. After drying for 1 h at 25 °C, the membrane was blocked with a blocking solution (1 % nonfat dry milk in DPBS) for 30 min at 25 °C. After washing once with a washing solution (0.05 % Tween 20 in DPBS), the membrane was washed twice with DPBS and dried for 1 h at RT. The conjugate pad was prepared by dispensing a desired volume of the conjugates and dried for 5 min at 25 °C. All components were assembled on a backing card and a strip cassette, respectively.
Fluorescence-based LFIA procedure and measurement method
LFIA was performed by applying 100 μL of the sample on a sample pad of the test strip. The capillary migration of the sample was allowed to process for 30 min in the dark at 25 °C. The test strip was inserted into a strip holder of the reader and the signal was measured at the excitation wavelength of 630 nm. Fluorescence intensity (FI) of the T spot and the C line were measured using a portable fluorescence lateral flow strip reader (Qiagen ESEQuant, Germany). The absolute fluorescence intensity (FIab) at the T spot was determined from the fluorescence intensity obtained from the sample containing the target NP (FIsample) subtracted with that of the negative control without the target (FIbuffer) as follows: (FIab) = (FIsample) - (FIbuffer).
Sensitivity and specificity
Various concentrations of rNP (0–1.5 μg·mL−1) were prepared before analysis in triplicate. To evaluate the sensitivity of the system, 100 μL of the prepared rNP samples were applied directly to the sample pad of the test strip and the signal was measured as described previously. The concentration-response curve was obtained by plotting the rNP concentration against the corresponding FIab value at the T spot. The detection limit was defined as the lowest concentration of rNP producing positive T spot FIab value, compared to the mean of negative controls plus 3 SD. DPBS was used as a negative control. Other proteins including avidin, lysozyme, BSA, tryptone, and amicase (5 μg·mL−1) were used for evaluation of the system specificity. A volume of 100 μL of each protein sample was applied directly to the test strip and processed in the same manner as described previously.
Detection of the NP antigen in the infected allantoic fluid
To evaluate the performance of the system for biological sample detection, the infected allantoic fluid was used as a sample and uninfected allantoic fluid was used as a negative control. The infected allantoic fluid was serially diluted with an extraction buffer (150 mM NaCl, 5 mM EDTA, 16 mM Triton X-100, 12 mM sodium deoxycholate, and 3.5 mM SDS in 10 mM phosphate buffer, pH 7.4) and incubated for 30 min at 4 °C. A volume of 100 μL of the treated allantoic fluid sample was applied directly to the sample pad. After 30-min incubation, the fluorescence signal at the T spot and C line was measured as described previously.
Results and discussion
Characterization of Cy5-doped silica nanoparticles and the conjugates
Cy5 fluorophores were covalently bound to organosilane through the reactive NHS ester group of Cy5-NHS ester and the amino group of APTES prior to particles formation. With this precoupling step, Cy5 dyes were efficiently encapsulated inside silica matrix [30] without leakage of dyes after several times of washing by both ethanol and water demonstrating high stability of dye-doped silica nanoparticle formation. Uniform and monodispersed nanoparticles with the size of 50 ± 6 (mean ± SD) nm were obtained (Fig. 1a). The maximum excitation and emission at 646 nm and 667 nm, respectively, of the particles were observed using spectrofluorometer (Fig. 1b). Compared to pure dye solution, which has the maximum excitation and emission at 643 nm and 662 nm, the spectrum of the particles showed small red-shift indicating that Cy5 dyes have not changed much after being encapsulated. It is worth noting that approximated number of dyes in the range of thousand molecules per particle has demonstrated in previous reports by comparison with calibration curves of pure dye solution [31]. In addition, Cy5-doped silica nanoparticle showed superior photostability to pure dyes demonstrating by He et al. [20]. The prepared particles were then modified with carboxyl silane in order to provide active carboxyl groups for further conjugation with targeting molecules. It has been described that adding unreactive phosphonate silane (THPMP) during the surface modification could reduce agglomeration and nonspecific binding of the particles even after conjugated with biomoieties [32]. Consequently, the mixture of carboxyl and phosphonate silane with the ratio of 1:1 was utilized for a post-coat step. To achieve specific targeting, monoclonal antibody specific to influenza A nucleoprotein was covalently conjugated to the modified particles through carbodiimide chemistry. The zeta potential of the particles was determined after each step of modification (Table S1, Electronic Supplementary Material; ESM). Plain particles exhibited negative zeta potential of −6.01 mV. After modification, carboxyl-modified particles became more negative (−14.7 mV) due to the presence of carboxyl groups on the surface. When MAb was covalently linked to the modified particles, less negative zeta potential value (−7.89 mV) of the conjugates was observed indicating the successful immobilization of antibodies to the particles.

Fluorescent dye-doped silica nanoparticles: a TEM image of Cy5-doped silica nanoparticles with the scale bar of 50 nm. b The excitation and emission spectra of the particles at 646 and 667 nm, respectively
Assay principle of fluorescence-based LFIA
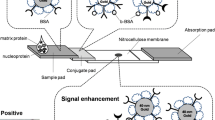
The principle of LFIA system using Cy5-doped silica nanoparticles as reporters is based on an integration of antibody-antigen specific recognition and high signal intensity of fluorescence-doped silica nanoparticles. Figure 2a illustrated the assay principle of fluorescence-based LFIA system. Within the system, the sample containing the target NP antigen migrated through a porous membrane by capillary action (Step 1). As the sample flowed along the device, the conjugates detected the NP antigen and formed complex (Step 2). The complex was then captured by the immobilized MAb on the T spot of the device and formed the sandwich immunocomplex. The accumulation of the sandwich complex enabled fluorescence signal detection of the particles. The excess conjugates migrated further and were captured by the secondary anti-mouse IgG at the C line indicated the validity of the test (Step 3). The presence of the fluorescence signal at both the T spot and the C line indicated the valid positive result, whilst the presence of the fluorescence signal at the C line only indicated the valid negative result. Quantitative analysis was achieved by obtaining the FIab value of the T spot using the portable reader (Fig. 2b).

Schematic diagram of (a) the fluorescence-based LFIA. The system components and principle of the test system were shown in Step 1. The NP antigen bound to the conjugates (Step 2) and flowed along the test strip to the T spot, where they were captured by the immobilized MAb to form a sandwich immunocomplex. The excess conjugates flowed further and captured by the secondary anti-mouse IgG Ab at the C line (Step 3). The accumulation of conjugates enabled the fluorescence signal detection, which was measured quantitatively using a portable strip reader. b Fluorescence images of valid positive and negative results (left panel) were shown with corresponding fluorescence intensity obtained from the reader (right panel)
Optimization of fluorescence-based LFIA system
The fluorescence signal response to this biosensor is directly related to the amount of the conjugates loaded on a conjugate pad since the measured signal depends on number of the conjugates captured at the T spot. Too low amount of the conjugates might limit sensitivity of the detection. In contrast, too high amount of the conjugates might generate high background resulting in a reduction of signal-to-noise (S/N) ratio. In order to obtain the optimal T spot, the concentration of the conjugates applied on the conjugate pad was optimized. The concentration of the conjugates was varied at 1, 5, 10 and 20 mg·mL−1, with applied volume of 2 μL. It was demonstrated that 10 mg·mL−1of the conjugates exhibited the maximum S/N ratio for samples containing 1.25 μg·mL−1of rNP, compared to that of other conjugate concentrations (Fig. S1, ESM). Consequently, 10 mg·mL−1 of the conjugates was used for the entire experiments. Further optimization to improve the signal strength at the T spot was also performed. The concentration of the free MAb immobilized at the T spot as a capture antibody was varied at 0.5, 1, and 2 mg·mL−1. It was observed that immobilizing free MAb at 1 and 2 mg·mL−1 produced maximum T spot signal, whilst MAb concentration of 0.5 mg·mL−1 generated relatively low signal due to too low amount of capture MAb availability (Fig. S2, ESM). However, 1 mg·mL−1of capture MAb was considered to be the best compromise between an economical use of antibody and the highest signal intensity. These optimal conditions were therefore adopted for all subsequent experiments.
Nonspecific interaction of nanoparticles is likely to limit the detection sensitivity by high generating background similar to most of other particle-based detection systems. To maximize the T spot signal and minimize fluorescence background, various formulas of the membrane blocking solution (1 % and 3 % of either BSA or nonfat dry milk) were prepared and evaluated. Figure 3 demonstrated that the T spot FIab value increased when the membrane was blocked with 1 % nonfat dry milk. Further investigation also showed that washing the blocked membrane with 0.05 % Tween 20 produced a stronger T spot signal (data not shown) than that of other washing solutions. Compared to the untreated membrane, the blocked membrane also showed a reduced background from nonspecific binding when evaluated with both the rNP and control, buffer only. However, reduced T spot FIab value was observed when high concentration (3 % BSA or 3 % nonfat dry milk) of blocking solution was used. The results prove that nonspecific binding has been significantly removed after blocking the membrane with blocking solution which may attribute to the shield effect of proteins from nonfat dry milk.

Comparison between unblocked (top), and blocked (bottom) membrane for the detection of (a) the rNP (1.25 μg·mL−1) and (b) negative control without rNP
Assay performance of fluorescence-based LFIA system
To demonstrate the performance of the system, the range of detection was determined by measuring fluorescence signal of samples with different rNP concentrations. The fluorescence signal at the T spot was recorded and the absolute fluorescence intensity (FIab) was calculated as mentioned above. The FIab was then plotted as a function of concentration of the rNP as shown in Fig. 4a. The result showed a continuous increase in FIab as the rNP concentration was raised from 0 to 1.5 μg·mL−1. Using this biosensor and a portable fluorescence strip reader, the rNP could be detected as low as 250 ng·mL−1 (25 ng of rNP per test) in one step. The low detection limit of this system can be attributed to the brightness of the particles containing thousands of dye molecules inside protective silica matrix. The superior brightness and photostability of the particles also allow the detection by a fluorescence imaging system (In-vivo MS FX Pro, Carestream Molecular Imaging, USA). An increase of the rNP concentration led to more accumulation of the conjugates at T spot, resulting in a growth in brightness of the fluorescence images, as shown in Fig. 4b. The bright image at the T spot was also observed at 250 ng·mL−1 of rNP target which corresponds to the detection limit using a portable strip reader.

a Evaluation of the fluorescence-based LFIA performance using various concentration of rNP. The rNP concentrations were plotted against the T spot fluorescence signal obtained from a portable reader after background subtraction with standard deviation. Arrows show the lowest detection limit of 250 ng·mL−1. Each concentration was performed in triplicate. b Images of the fluorescence-based LFIA test strips evaluated with different concentration of rNP (0–1.5 μg·mL−1)
To determine whether the detection limit of the fluorescence-based LFIA is comparable to other immunoassay based systems; a number of studies were reviewed for data reporting a detectable level of the influenza NP using different approaches. The previous studies reported various detectable level of the influenza A NP. For example, Jianumpunkul et al. reported 200 ng of the rNP using triple-antigen sandwich ELISA (equivalent to 4 μg·mL−1 for comparison) [33]. Farris et al. detected 1 μg·mL−1of the total protein of influenza A virus using immuno-interferometric method [34]. A lower limit of detection of the rNP (10 ng·mL−1) has also been reported using electrochemical immunoassay [35]. Compared to the methods listed above, it was shown that our system exhibited lower detection limit than that of the ELISA and immuno-interferometric assays. Although electrochemical immunoassay provided superior sensitivity, complicated and costly system is required. The differences in a detection limit of the influenza A NP from each individual approach could be contributed to a number of factors. These, for example, included the affinity of targeting ligands (antibody), the antigen type (recombinant protein or inactivated virus), labeling molecules, detection signal, and the optimal conditions used in each individual approach.
To assess the specificity of detection, the fluorescence intensity was measured within 30 min after applying a sample containing the target rNP (1.25 μg·mL−1) or other proteins, such as BSA, avidin, lysozyme, tryptone, and amicase (5 μg·mL−1). As shown in Fig. 5, strong fluorescence signal of a sample containing rNP was observed, while the other proteins showed very low signal as background. The result demonstrated high rNP specificity of this biosensor against other proteins, which may have otherwise interfered with detection in biological samples.

Specificity of the fluorescence-based LFIA. The system was successfully used to detect the rNP (1.25 μg·mL−1) as demonstrated by the corresponding FIab value. The system showed negative results when evaluated with other proteins (5 μg·mL−1). Error bars represent standard deviation (n = 3)
Detection of the NP in infected allantoic fluid
To demonstrate the feasibility of using fluorescence-based LFIA for influenza A antigen detection in complex biological matrices, the system was applied to detect NP in influenza A-infected allantoic fluid directly. Pandemic 2009 influenza A H1N1 virus-infected allantoic fluid stock solution containing 5.6 × 103 TCID50·mL−1 (50 % tissue culture infectious dose) was used as a sample. It should be noted that the stock solution of the infected allantoic fluid used in this study was prepared using the early passage pandemic 2009 influenza A virus. This would therefore result in a low titer of the virus. Prior to a sample application, the infected allantoic fluid was treated with an extraction buffer in order to lyse the virus particles. Two-fold serial dilution of the infected allantoic fluid was prepared and applied to the system. Similar to the rNP, the T spot FIab was shown to be increased when the concentration of the infected allantoic fluid was increased from 0 to 2.8 × 103 TCID50·mL−1as demonstrated in Fig. 6. It was also shown that the system was able to detect NP in the infected allantoic fluid with the lowest dilution of 1.1 × 101 TCID50·mL−1 (FIab = 98 ± 14 a.u.). Nevertheless, uninfected allantoic fluid used as a negative control showed no T spot signal indicating neither nonspecific binding nor false positive signal. The highly specific and sensitive detection of the target analytes in allantoic fluid might be attributed to a low background noise when the wavelength in red spectral region was used to exclude the intrinsic fluorescence of most biological samples and the scatter of nitrocellulose membrane as mentioned previously. This indicated that the developed system could be potentially used to detect the target analytes in complex biological samples without background noise that could interfere with the system functionality (as shown by the T spot and C line signal development).

a Analytical performance of the fluorescence-based LFIA in detecting NP in the allantoic fluid infected with pandemic influenza A H1N1 virus. The infected allantoic fluid was 2-fold serially diluted and plotted against the FIab value obtained from a portable reader after background subtraction. Uninfected allantoic fluid was used as negative control. Each concentration was performed triplicately. b Images of the fluorescence-based LFIA test strips evaluated by the infected allantoic fluid. Arrows showed the lowest detection limit of 1.1 × 101 TCID50·mL−1
Further investigation to compare the system performance with a conventional LFIA system using gold nanoparticles as labels for colorimetric detection was performed. It was shown that the commercial Flu A test based on conventional LFIA system could detect NP in the infected allantoic fluid with the LOD of 8.8 × 101 TCID50·mL−1 (Fig. S3, ESM). Compared to the conventional test, the fluorescence-based LFIA system exhibited high sensitivity, as illustrated by the eight-fold lower detection limit than that of the commercial one. Overall results demonstrated that, the developed system using Cy5-doped silica nanoparticles as reporters could potentially become an alternative system for target analyte detection, not only due to its simplicity but also good sensitivity.
Conclusion
A fluorescence-based LFIA using Cy5-doped silica nanoparticles as labels was successfully developed for a rapid and sensitive detection of the target influenza A NP. The bright fluorescence signal could be achieved proportionally to the NP concentration and the system was capable of detecting the target protein in a nanogram range. Furthermore, the fluorescence signal could be directly measured using a portable strip reader allowing rapid and quantitative analysis. This system could also successfully detect the NP in biological matrices such as infected allantoic fluid without background interferences from sample matrix components. This study demonstrated a proof of concept in employing Cy5-doped silica nanoparticles as labels for fluorescence-based LFIA. The system performed sensitive and one-step quantitative detection of the target analytes which could be applied in many different medical and diagnostic applications.
References
Porstmann T, Kiessig ST (1992) Enzyme immunoassay techniques: an overview. J Immunol Methods 24:5–21
Kurita R, Arai K, Nakamoto K, Kato D, Niwa O (2010) Development of electrogenerated chemiluminescence-based enzyme linked immunosorbent assay for sub-pM detection. Anal Chem 82:1692–1697
Wingren C, Borrebaeck CA (2009) Antibody microarrays: current status and key technologies advances. OMICS 10:1692–1697
Wang Z, Lee J, Cossins AR, Brust M (2005) Microarray-based detection of protein binding and functionality by gold nanoparticle probes. Anal Chem 77:5770–5774
Meyer MHF, Hartmann M, Keusgen M (2006) SPR-based immunosensor for the CRP detection-A new method to detect a well known protein. Biosens Bioelectron 21:1987–1990
Temur E, Zengin A, Boyaci IH, Dudak FC, Torul H, Tamer U (2012) Attomole sensitivity of staphylococcal enterotoxin B detection using an aptamer-modified surface-enhanced Raman scattering probe. Anal Chem 84:10600–10606
Yonzon CR, Haynes CL, Zhang X, Walsh JT, Van Duyne RP (2004) A glucose biosensor based on surface-enhanced Raman scattering: improved partition layer, temporal stability, reversibility, and resistance to serum protein interference. Anal Chem 76:78–85
Posthuma-Trumpie GA, Korf J, van Amerogen A (2009) Lateral flow (immuno) assay: its strengths, weaknesses, opportunities and threats: a literature survey. Anal Bioanal Chem 393:569–582
Xu H, Mao X, Zeng Q, Wang S, Kawde AN, Liu G (2009) Aptamer-functionalized gold nanoparticles as probes in a dry-reagent strip biosensor for protein analysis. Anal Chem 81:669–675
Mizuike R, Sasaki T, Baba K, Iwamoto H, Shibai Y, Kosaka M, Kubota-Koketsu R, Yang CS, Du A, Sakudo A, Tsujikawa M, Yunoki M, Ikuta K (2011) Development of two types of rapid diagnostic test kits to detect the hemagglutinin or nucleoprotein of the swine-origin pandemic influenza A virus H1N1. Clin Vaccine Immunol 18:494–499
Wiriyachaiporn S, Howarth PH, Bruce KD, Dailey LA (2013) Evaluation of a rapid lateral flow immunoassay for Staphylococcus aureus detection in respiratory samples. Diagn Microbiol Infect Dis 75:28–36
Paek SH, Lee SH, Cho JH, Kim YS (2000) Development of rapid one-step immunochromatographic assay. Methods 22:53–60
Choi DH, Lee SK, Oh YK, Bae BW, Lee SD, Kim S, Shin YB, Kim MG (2010) A dual gold nanoparticle conjugate-based lateral flow assay (LFA) method for the analysis of troponin I. Biosens Bioelectron 15:1999–2002
Khreich N, Lamourette P, Boutal H, Devilliers K, Créminon C, Volland H (2008) Detection of Staphylococcus enterotoxin B using fluorescent immunoliposomes as label for immunochromatograhic testing. Anal Biochem 377:182–188
O’Keeffe M, Crabbe P, Salden M, Wichers J, Van Peteghem C, Kohen F, Pieraccini G, Moneti G (2003) Preliminary evaluation of a lateral flow immunoassay device for screening urine samples for the presence of sulphamethazine. J Immunol Methods 278:117–126
Kim YM, Oh SW, Jeong SY, Pyo DJ, Choi EY (2003) Development of an ultrarapid one-step fluorescence immunochromatographic assay system for the quantification of microcystins. Environ Sci Technol 37:1899–1904
Ahna JS, Choia S, Janga SH, Changb HJ, Kimc JH, Nahmc KB, Oha SW, Choi EY (2003) Development of a point-of-care assay system for high-sensitivity C-reactive protein in whole blood. Clin Chim Acta 332:51–59
Bae SW, Tan W, Hong JI (2012) Fluorescent dye-doped silica nanoparticles: new tools for bioapplications. Chem Commun 48:2270–2282
Fare TL, Coffey EM, Dai HY, He YDD, Kessler DA, Kilian KA, Koch JE, LeProust E, Marton MJ, Meyer M, Stoughton RB, Tokiwa GY, Wang YQ (2003) Effect of atmospheric ozone on microarray data quality. Anal Chem 75:4672–4675
He X, Chen J, Wang K, Qin D, Tan W (2007) Preparation of luminescent Cy5 doped core-shell SFNPs and its application as near-infrared fluorescent marker. Talanta 72:1519–1526
Brigg MS, Burns DD, Cooper ME, Gregory SJ (2000) A pH sensitive fluorescent cyanine dye for biological applications. Chem Commun 23:2323–2324
Tan W, Wang K, He X, Zhao XJ, Drake T, Wang L, Bagwe RP (2004) Bionanotechnology based on silica nanoparticles. Med Res Rev 24:621–638
Santra S, Zhang P, Wang K, Tapec R, Tan W (2001) Conjugation of biomolecules with luminophore-doped silica nanoparticles for photostable biomarkers. Anal Chem 73:4988–4993
Smith JE, Medley CD, Tang Z, Shangguan D, Lofton C, Tan W (2007) Aptamer-conjugated nanoparticles for the collection and detection of multiple cancer cells. Anal Chem 79:3075–3082
Watcharatanyatip K, Boonmoh S, Chaichoun K, Songserm T, Woratanti M, Dharakul T (2010) Multispecies detection of antibodies to influenza A viruses by a double-antigen sandwich ELISA. J Virol Methods 163:238–243
World Health Organization (2002) WHO manual on animal influenza diagnosis and surveillance. http://www.who.int/vaccine_research/diseases/influenza/WHO_manual_on_animal-diagnosis_and_surveillance_2002_5.pdf Accessed 10 April 2013
King DJ (1991) Evaluation of different methods of inactivation of newcastle disease virus and avian influenza virus in egg fluids and serum. Avian Dis 35:505–514
Barry RD, Davies P (1968) The sedimentation of influenza virus and its RNA in sucrose density gradients. J Gen Virol 2:59–69
Stober W, Fink A, Bohn E (1968) Controlled growth of monodisperse silica spheres in the micron size range. J Colloid Interface Sci 26:62–69
Wang L, Tan W (2006) Multicolor FRET silica nanoparticles by single wavelength excitation. Nano Lett 6:84–88
Estévez MC, O’Donoghue MB, Chen X, Tan W (2009) Highly fluorescent dye-doped silica nanoparticles increase flow cytometry sensitivity for cancer cell monitoring. Nano Res 2:448–461
Medley CD, Bamrungsap S, Tan W, Smith JE (2011) Aptamer-conjugated nanoparticles for cancer cell detection. Anal Chem 83:727–734
Jian-umpunkul P, Thepthai C, Apiwat N, Chantima W, Poomputsa K, Wiriyachaiporn N, Dharakul T (2012) Improved sensitivity of influenza A antigen detection using a combined NP, M, and NS1 sandwich ELISA. J Virol Methods 185:24–31
Farris LR, Wu N, Wang W, Clarizia LA, Wang X, McDonald MJ (2010) Immuno-interferometric sensor for the detection of influenza A nucleoprotein. Anal Bioanal Chem 396:667–674
Ohtsuka K, Endo H, Morimoto K, Vuong BN, Ogawa H, Imai K, Takenaka S (2008) Detection of an antibody to avian influenza virus by an electrochemical immunoassay (eELISA). Anal Sci 24:1619–1622
Acknowledgments
This work was supported by grant from NANOTEC, Thailand.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 161 kb)
Rights and permissions
About this article
Cite this article
Bamrungsap, S., Apiwat, C., Chantima, W. et al. Rapid and sensitive lateral flow immunoassay for influenza antigen using fluorescently-doped silica nanoparticles. Microchim Acta 181, 223–230 (2014). https://doi.org/10.1007/s00604-013-1106-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-013-1106-4