
Abstract
Background
Alport syndrome (AS), atypical hemolytic-uremic syndrome (aHUS), and fibronectin-glomerulopathy (FG) are rare forms of glomerular diseases that manifest in a combination of proteinuria, hematuria, and hypertension, referred to as nephritic syndrome. Due to phenotypic overlays, steroid-resistant nephrotic syndrome (SRNS) and nephritic syndrome have been difficult to discern diagnostically. SRNS is more common than nephritic syndrome and is the second leading cause of childhood-onset CKD. Fourteen monogenic causes of AS, aHUS, and FG and 60 monogenic causes of SRNS have been identified. As whole exome sequencing (WES) allows for unequivocal molecular genetic diagnostics, we hypothesize to be able to identify causative mutations in genes known to cause nephritic syndrome in patient cohorts with a clinical diagnosis of SRNS.
Methods
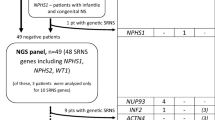
We identified patients with hematuria and steroid-resistant proteinuria in an international patient cohort that we had submitted to WES and who were unsolved for known monogenic causes of SRNS. These 70 patients from 65 individual families were subsequently analyzed for causative mutations in 14 AS, aHUS, or FG causing genes. WES data were compared to a control cohort of 76 patients from 75 families that were diagnosed with nephronophthisis-related ciliopathies (NPHP-RC) and to a control cohort of 83 individuals from 75 families with SRNS, but without hematuria.
Results
We detected likely pathogenic genetic variants in 3 of 65 families (4.6%) in 2 of the 14 genes analyzed.
Conclusions
We confirmed that in cohorts of childhood-onset SRNS, patients with nephritic syndrome can be discerned by WES. The findings highlight the importance of clinical genetic testing for therapeutic and preventative measures in patients with proteinuria.
Graphical abstract
A higher resolution version of the Graphical abstract is available as Supplementary information.

Similar content being viewed by others


Data availability
The data will be shared on reasonable request made to the corresponding author.
References
Kopp JB, Anders HJ, Susztak K et al (2020) Podocytopathies. Nat Rev Dis Primers 6:1–24. https://doi.org/10.1038/s41572-020-0196-7
Lovric S, Ashraf S, Tan W, Hildebrandt F (2016) Genetic testing in steroid-resistant nephrotic syndrome: when and how? Nephrol Dial Transplant 31:1802–1813. https://doi.org/10.1093/ndt/gfv355
Lamba P, Nam KH, Contractor J, Kim A (2020) Nephritic syndrome. Prim Care 47:615–629. https://doi.org/10.1016/j.pop.2020.08.003
Nozu K, Nakanishi K, Abe Y et al (2019) A review of clinical characteristics and genetic backgrounds in Alport syndrome. Clin Exp Nephrol 23:158–168. https://doi.org/10.1007/s10157-018-1629-4
Lemmink HH, Mochizuki T, van den Heuvel LP et al (1994) Mutations in the type IV collagen alpha 3 (COL4A3) gene in autosomal recessive Alport syndrome. Hum Mol Genet 3:1269–1273
Mochizuki T, Lemmink HH, Mariyama M et al (1994) Identification of mutations in the alpha 3(IV) and alpha 4(IV) collagen genes in autosomal recessive Alport syndrome. Nat Genet 8:77–81. https://doi.org/10.1038/ng0994-77
Antignac C, Knebelmann B, Drouot L et al (1994) Deletions in the COL4A5 collagen gene in X-linked Alport syndrome. Characterization of the pathological transcripts in nonrenal cells and correlation with disease expression. J Clin Invest 93:1195–1207. https://doi.org/10.1172/JCI117073
Zhang Y, Bockhaus J, Wang F et al (2021) Genotype-phenotype correlations and nephroprotective effects of RAAS inhibition in patients with autosomal recessive Alport syndrome. Pediatr Nephrol 36:2719–2730. https://doi.org/10.1007/s00467-021-05040-9
Vivante A, Hildebrandt F (2016) Exploring the genetic basis of early-onset chronic kidney disease. Nat Rev Nephrol 12:133–146. https://doi.org/10.1038/nrneph.2015.205
Lemaire M, Fremeaux-Bacchi V, Schaefer F et al (2013) Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet 45:531–536. https://doi.org/10.1038/ng.2590
Neumann HP, Salzmann M, Bohnert-Iwan B et al (2003) Haemolytic uraemic syndrome and mutations of the factor H gene: a registry-based study of German speaking countries. J Med Genet 40:676–681
Noris M, Caprioli J, Bresin E et al (2010) Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 5:1844–1859. https://doi.org/10.2215/CJN.02210310
Westra D, Vernon KA, Volokhina EB, Pickering MC, van de Kar NC, van den Heuvel LP (2012) Atypical hemolytic uremic syndrome and genetic aberrations in the complement factor H-related 5 gene. J Hum Genet 57:459–464. https://doi.org/10.1038/jhg.2012.57
Smith JM, Stablein DM, Munoz R, Hebert D, McDonald RA (2007) Contributions of the Transplant Registry: The 2006 Annual Report of the North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS). Pediatr Transplant 11:366–373. https://doi.org/10.1111/j.1399-3046.2007.00704.x
Fakhouri F, Zuber J, Frémeaux-Bacchi V, Loirat C (2017) Haemolytic uraemic syndrome. Lancet 390:681–696. https://doi.org/10.1016/s0140-6736(17)30062-4
Nakapoulou I, Stefanaki K, Zeis PM et al (1993) The glomerular distribution of laminin and fibronectin in glomerulonephritis. Histol Histopathol 8:521–526
Castelletti F, Donadelli R, Banterla F et al (2008) Mutations in FN1 cause glomerulopathy with fibronectin deposits. Proc Natl Acad Sci U S A 105:2538–2543. https://doi.org/10.1073/pnas.0707730105
Schapiro D, Daga A, Lawson JA et al (2019) Panel sequencing distinguishes monogenic forms of nephritis from nephrosis in children. Nephrol Dial Transplant 34:474–485. https://doi.org/10.1093/ndt/gfy050
Connaughton DM, Kennedy C, Shril S et al (2019) Monogenic causes of chronic kidney disease in adults. Kidney Int 95:914–928. https://doi.org/10.1016/j.kint.2018.10.031
Warejko JK, Tan W, Daga A et al (2018) Whole exome sequencing of patients with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 13:53–62. https://doi.org/10.2215/cjn.04120417
Sherry ST, Ward MH, Kholodov M et al (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29:308–311. https://doi.org/10.1093/nar/29.1.308
Braun DA, Sadowski CE, Kohl S et al (2016) Mutations in nuclear pore genes NUP93, NUP205 and XPO5 cause steroid-resistant nephrotic syndrome. Nat Genet 48:457–465. https://doi.org/10.1038/ng.3512
van der Ven AT, Connaughton DM, Ityel H et al (2018) Whole-exome sequencing identifies causative mutations in families with congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol 29:2348–2361. https://doi.org/10.1681/asn.2017121265
Hildebrandt F, Heeringa SF, Ruschendorf F et al (2009) A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS Genet 5:e1000353. https://doi.org/10.1371/journal.pgen.1000353
Sayer JA, Otto EA, O’Toole JF et al (2006) The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet 38:674–681
Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC (2012) SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res 40:W452-457. https://doi.org/10.1093/nar/gks539
Schwarz JM, Cooper DN, Schuelke M, Seelow D (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11:361–362. https://doi.org/10.1038/nmeth.2890
Adzhubei IA, Schmidt S, Peshkin L et al (2010) A method and server for predicting damaging missense mutations. Nat Methods 7:248–249. https://doi.org/10.1038/nmeth0410-248
Karczewski KJ, Francioli LC, Tiao G et al (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581:434–443. https://doi.org/10.1038/s41586-020-2308-7
Stenson PD, Mort M, Ball EV et al (2020) The Human Gene Mutation Database (HGMD(®)): optimizing its use in a clinical diagnostic or research setting. Hum Genet 139:1197–1207. https://doi.org/10.1007/s00439-020-02199-3
Landrum MJ, Chitipiralla S, Brown GR et al (2020) ClinVar: improvements to accessing data. Nucleic Acids Res 48:D835–D844. https://doi.org/10.1093/nar/gkz972
Yates AD, Achuthan P, Akanni W et al (2020) Ensembl 2020. Nucleic Acids Res 48:D682–D688. https://doi.org/10.1093/nar/gkz966
Sievers F, Wilm A, Dineen D et al (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. https://doi.org/10.1038/msb.2011.75
Delvaeye M, Noris M, De Vriese A et al (2009) Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med 361:345–357. https://doi.org/10.1056/NEJMoa0810739
Edelsten AD, Tuck S (1978) Familial haemolytic uraemic syndrome. Arch Dis Child 53:255–256
Fremeaux-Bacchi V, Dragon-Durey MA, Blouin J et al (2004) Complement factor I: a susceptibility gene for atypical haemolytic uraemic syndrome. J Med Genet 41:e84
Fremeaux-Bacchi V, Miller EC, Liszewski MK et al (2008) Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood 112:4948–4952. https://doi.org/10.1182/blood-2008-01-133702
Gale DP, de Jorge EG, Cook HT et al (2010) Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet 376:794–801. https://doi.org/10.1016/S0140-6736(10)60670-8
Levy GG, Nichols WC, Lian EC et al (2001) Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature 413:488–494. https://doi.org/10.1038/35097008
Noris M, Brioschi S, Caprioli J et al (2003) Familial haemolytic uraemic syndrome and an MCP mutation. Lancet 362:1542–1547. https://doi.org/10.1016/S0140-6736(03)14742-3
Venables JP, Strain L, Routledge D et al (2006) Atypical haemolytic uraemic syndrome associated with a hybrid complement gene. PLoS Med 3:e431. https://doi.org/10.1371/journal.pmed.0030431
Matthaiou A, Poulli T, Deltas C (2020) Prevalence of clinical, pathological and molecular features of glomerular basement membrane nephropathy caused by COL4A3 or COL4A4 mutations: a systematic review. Clin Kidney J 13:1025–1036. https://doi.org/10.1093/ckj/sfz176
Torra R, Furlano M, Ars E (2020) How genomics reclassifies diseases: the case of Alport syndrome. Clin Kidney J 13:933–935. https://doi.org/10.1093/ckj/sfaa170
Voskarides K, Damianou L, Neocleous V et al (2007) COL4A3/COL4A4 mutations producing focal segmental glomerulosclerosis and renal failure in thin basement membrane nephropathy. J Am Soc Nephrol 18:3004–3016. https://doi.org/10.1681/asn.2007040444
Jayasinghe K, Stark Z, Kerr PG et al (2021) Clinical impact of genomic testing in patients with suspected monogenic kidney disease. Genet Med 23:183–191. https://doi.org/10.1038/s41436-020-00963-4
Pinto EVF, Kemppainen JL, Lieske JC, Harris PC, Hogan MC (2021) Establishing a nephrology genetic clinic. Kidney Int 100:254–259. https://doi.org/10.1016/j.kint.2021.05.008
Acknowledgements
We thank the participating families and the physicians for their contributions. F.H. is the William E. Harmon Professor of Pediatrics
Funding
This research was supported by grants from the National Institutes of Health (5R01DK076683-15 to FH).
Author information
Authors and Affiliations
Contributions
HX: analysis and interpretation of data, drafting of article; FH: conception and design of research work, drafting and revision of article.
Corresponding author
Ethics declarations
Ethics approval
This study was approved by the institutional review boards of Boston Children’s Hospital and the University of Michigan. DNA samples were collected between 09/1996 and 04/2016 for the nephritis cohort, and between 09/2017 and 08/2019 for the NPHP-RC cohort, after obtaining written informed consent, clinical data, and pedigree information (www.renalgenes.org).
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xiao, H., Hildebrandt, F. Whole exome sequencing identifies monogenic forms of nephritis in a previously unsolved cohort of children with steroid-resistant nephrotic syndrome and hematuria. Pediatr Nephrol 37, 1567–1574 (2022). https://doi.org/10.1007/s00467-021-05312-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-021-05312-4