
Abstract
Otofaciocervical syndrome (OFCS) is an autosomal recessively inherited disorder characterized by facial dysmorphism, external ear anomalies with preauricular pits and hearing impairment, branchial cysts or fistulas, anomalies of the vertebrae and the shoulder girdle, and mild intellectual disability. In a large consanguineous family with OFCS from Turkey, we performed whole-exome sequencing (WES) of a single pooled DNA sample of four affected individuals. Filtering for variants with a percentage of alternate reads ≥90 % and a coverage of at least five reads identified only a single novel homozygous variant, c.497G>T, located in PAX1 that co-segregated with the disease in the family. PAX1 encodes a transcription factor with a critical role in pattern formation during embryogenesis in vertebrates. The mutation is predicted to substitute the glycine at position 166 to valine (p.G166V) within the highly conserved paired-box domain of the PAX1 protein. We performed a dual luciferase reporter assay to examine the transactivation of a regulatory sequence in the Nkx3-2 promoter region, which is a direct target of mouse Pax1 transcriptional regulation. We observed a significantly reduced transactivation in HEK293T cells overexpressing Pax1G157V in comparison to Pax1WT expressing cells, indicating a reduced DNA-binding affinity of the mutant protein. Taken together, our results show that the strategy of pooling DNA is a powerful, cost-effective application for WES in consanguineous families and establish PAX1 as a new disease-causing gene for OFCS and as part of the EYA-DACH-SIX-PAX network, important in early embryogenesis.
Similar content being viewed by others

Introduction
Otofaciocervical syndrome (OFCS, MIM 166780) is a rare disorder characterized by facial anomalies, cup-shaped low-set ears, preauricular fistulas, hearing loss, branchial defects, skeletal anomalies including vertebral defects, low-set clavicles, winged scapulae, sloping shoulders, and mild intellectual disability. The disorder was first described by Fara et al. (1967) in an affected father and four of his seven children. Apart from the skeletal anomalies and absence of renal abnormalities, OFCS is clinically very similar to branchiootorenal syndrome (BORS, MIM 113650), and sometimes its distinction from BORS is difficult. Mutations in EYA1 account for nearly 70 % of cases with BORS (Abdelhak et al. 1997a, b; Rickard et al. 2000) and, interestingly, mutations in EYA1 could also be identified in patients with OFCS (Rickard et al. 2001; Estefania et al. 2005). BORS is part of the branchial-arch spectrum disorders, generally characterized by ear malformations, branchial cleft sinuses, mixed-type hearing loss, and renal anomalies. This spectrum also includes the branchiootic syndrome (BOS, MIM 602588) and the branchiooculofacial syndrome (BOFS, MIM 113620), which additionally shows eye involvement and intellectual disability. Identified mutations in these overlapping syndromes established the EYA-DACH-SIX-PAX pathway, a molecular network involved in the pathogenesis of branchial-arch spectrum disorders (Abdelhak et al. 1997a, b; Ruf et al. 2004; Kaiser et al. 2007).
The PAX gene family is a highly conserved group of developmental control genes, which encode transcription factors with a role in pattern formation during vertebrate embryogenesis (Chi and Epstein 2002; Read and van Heyningen 1994; Noll 1993). To date, nine human paired-box genes have been described and divided into four distinct groups according to their protein structure (Lang et al. 2007). Specifically, PAX1 is expressed during the development of the skeleton, thymus, and parathyroid glands (Schnittger et al. 1992; Wallin et al. 1996) and is required for normal development of ventral vertebral structures (Wallin et al. 1994). Interestingly, previous studies could identify Nkx3-2 (NK3 homeobox 2; Bapx1), which is expressed in the embryonic tissue of the sclerotome and in developing vertebrae, as a target of Pax1 and Pax9 transcriptional activation (Rodrigo et al. 2003). We show that a homozygous missense mutation in PAX1 (NM_006192.4) causes OFCS in four affected family members of a large consanguineous family from Turkey. The mutation leads to a significantly reduced transactivation of a regulatory sequence of the Nkx3-2 promoter, providing evidence for the hypofunctional nature of the mutation.
Materials and methods
Human subjects
All subjects or their legal representatives gave written informed consent to the study. The study was performed in accordance to the Declaration of Helsinki protocols and approved by the local institutional review board of the Medical Faculty at Ege University, Izmir, Turkey. DNA from participating family members was extracted from peripheral blood lymphocytes by standard extraction procedures.
Next generation sequencing
Equal amounts of genomic DNA of the four affected individuals IV-9, IV-10, V-2, and V-3 were pooled (0.75 μg DNA per sample). Exonic and adjacent intronic sequences were enriched from the single “mixed” genomic DNA of the affected individuals with the use of the Agilent SureSelect Human Exome Kit V2 and run on the Illumina Genome Analyzer IIX Sequencer by Oxford Gene Technology. The reads were aligned to the human reference genome (human_g1k_v37) using Burrows-Wheeler Aligner (BWA v0.6.2), and local realignment around indels, base quality recalibration and variant calling were performed with Genome Analysis Toolkit (GATK v2.1). The resulting variants were annotated with Annovar (v2012oct23) and filtered according to their genomic position, predicted effect and frequency in the general population, as found in dbSNP 135, the 1000 Genomes Project, the exome variant server and our in-house database, which included 43 unrelated samples.
Genetic analysis
The identified mutation was re-sequenced by Sanger sequencing in an independent experiment and tested for co-segregation with the disease within the family. Haplotype analysis was performed using polymorphic microsatellite markers to confirm the critical homozygous region including the PAX1 gene. Primer sequences to amplify specific markers were taken from ENSEMBL. For fine mapping of the PAX1 region surrounded by critical recombination breakpoints in two affected individuals, additional polymorphic markers were identified with the UCSC Genome Browser hg19 and primers were designed. The following polymorphic regions on chromosome 20 were amplified and used for marker analysis: D20S432 (19,831,622–19,831,792), D20S912 (20,860,834–20,861,112), D20S54 (20,943,970–20,944,161), D20S63 (21,000,111–21,000,227), D20S_PAX1.1 (21,592,785–21,592,821), D20S_PAX1.3 (21,686,081–21,686,124), D20S_PAX1.4 (21,969,695–21969731), D20S477 (22,412,084–22,412,342), D20S871 (23,335,573–23,335,756), D20S148 (23,605,303–23,605,643). D20S_PAX1.1, D20S_PAX1.3, and D20S_PAX1.4 were originally designed. Ten variations detected in the exome data set with a percentage of alternate reads between 75 and 80 % were randomly chosen and re-sequenced by Sanger sequencing in all affected family members.
Generation of Nkx3-2 and Pax1 constructs
A regulatory sequence in the mouse Nkx3-2 promoter region (5′ end position at −880, 3′ end position +109; +1 as first nucleotide in GenBank cDNA U87957.1; Rodrigo et al. 2003) as a direct target of Pax1 transcriptional regulation was amplified and cloned into the firefly reporter plasmid pGL4.10 (Promega). To generate Pax1 expression plasmids containing the mouse Pax1 WT or Pax1 G157V coding sequences (NM_008780.2) (the glycine residue at position 157 in the mouse Pax1 protein is the corresponding amino acid to the glycine at position 166 in human PAX1), the coding sequence of mouse Pax1 with an additional HA tag was amplified by RT-PCR from isolated adult mouse thymus RNA and cloned into the mammalian expression vector pEGFPN1∆EGFP. The mutant variant of Pax1 was generated by site-directed mutagenesis with primers containing the specific nucleotide substitution. The cDNA sequences of the constructs were confirmed by Sanger sequencing.
Luciferase assay
HEK293T cells cultured in Dulbecco’s modified Eagle media (DMEM) containing 10 % fetal bovine serum (FBS, Life Technologies) and antibiotics were plated in 24-well plates 18 h prior to transfection. Transient transfections were performed in triplicate with FuGeneHD (Roche) according to the manufacturer’s instructions. Cells were cotransfected with 200 ng of expression vector containing HA-tagged Pax1 WT or Pax1 G157V, 100 ng of firefly reporter plasmid Nkx3-2-pGL4.10 and 5 ng of pRL-TK vector for normalization. To ensure equal amounts of transfected DNA and to determine the baseline activity, 200 ng LacZ-pcDNA3.1 was used in Pax1 negative transfections. After 30 h of transfection, cell extracts were collected and frozen in lysis buffer overnight at −20 °C to increase the lysing efficiency. After thawing, firefly and renilla luciferase activities were measured using a Dual Luciferase Reporter Assay Kit (Promega) and a Glomex 96-microplate luminometer. To correct for variations in transfection efficiency, firefly luciferase activity was normalized to renilla luciferase. Three experiments were performed in triplicate.
Western blot analysis
To test the expression levels of Pax1WT and mutant Pax1G157V proteins after measurement of the luciferase activity, 5 μl of whole cell lysate per sample was subjected to reducing 4–12 % SDS-PAGE (Invitrogen) and transferred to a nitrocellulose membrane. Immunoblots were blocked in 5 % milk powder in TBS containing 0.1 % Tween 20 and probed with anti-HA high affinity antibody (Roche). Equal amounts of protein were confirmed by β-actin (Sigma) detection. Peroxidase conjugated secondary antibodies were purchased from Santa Cruz and blots were developed using an enhanced chemiluminescence system, Super Signal West Pico Chemiluminescent Substrate (Thermo Fisher), followed by exposure on an autoradiographic film (GE Healthcare).
Expression analysis in mouse cochlea
Cochleae from P6 mice were dissected and transferred in RNAlater RNA Stabilization Reagent (Qiagen). After homogenization, RNA was isolated using the Trizol (Life Technologies) method. Total cDNA was transcribed by RevertAid First Strand cDNA synthesis Kit (Thermo Scientific) using random hexamer primers and used for Pax1-specific PCR amplification. PCR primers for RT-PCR were located in exon 3 (forward, 5′-CCAGCAGTGAATGGACTCG-3′) and exon 5 (reverse, 5′-AGGTCGAGGCTGGGATG-3′) of mouse Pax1. PCR products were separated on a 1 % agarose gel and verified by Sanger sequencing.
Web resources
-
UCSC browser (hg19), http://genome.ucsc.edu/index.html;
-
ENSEMBL, http://www.ensembl.org; NCBI, http://www.ncbi.nlm.nih.gov/;
-
UniProt, http://www.uniprot.org/; OMIM, http://www.ncbi.nlm.nih.gov/omim;
-
The exome variant server (ESP6500SI-V2), http://evs.gs.washington.edu/EVS/;
-
Pfam, http://pfam.sanger.ac.uk/search?tab=searchSequenceBlock;
-
SMART, http://smart.embl-heidelberg.de/smart/set_mode.cgi?NORMAL=1;
-
BLAST, http://blast.ncbi.nlm.nih.gov/;
-
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/;
-
ConSeq, http://conseq.tau.ac.il/;
-
MutationTaster, http://www.mutationtaster.org/.
Results
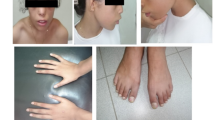
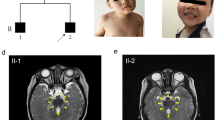
We have examined a large consanguineous Turkish family with autosomal recessively inherited OFCS in four affected family members from two different branches of the pedigree (Fig. 1a; Table 1). The index patient V-2 was referred to the Pediatric Genetics Subdivision at Ege University, Izmir, Turkey, because of the unusual face and mild developmental delay. All affected individuals in the family presented with cup-shaped ears, bilateral mixed hearing loss, bilateral preauricular fistulas, lacrimal duct abnormalities, protruding shoulders, and winged scapulae. The index patient had normal birth weight and height, but showed moderately delayed motor and mental milestones. On physical examination, he showed facial features which included long eyelashes, blue sclera, downslanting palpebral fissures, bilateral hypoplastic lacrimal puncta, bilateral preauricular fistulas, cup-shaped and low-set ears, a mixed type of hearing loss (65 dB left and 75 dB right hearing threshold), and microretrognathia. Intra-oral examination revealed malocclusion and dental caries. He had protruding shoulders, scapular winging, tapering fingers and bilateral clinodactyly. There was bilateral cutaneous syndactyly of the second and third toes (Fig. 1b, c). After excision of a preauricular fistula, pathological examination showed that the fistula was originated from the branchial arch. Bone X-rays revealed anterior scalloping of the lower thoracic and lumbar vertebral bodies and a fusion defect in L5/S1. Cranial magnetic resonance imaging showed periventricular white matter gliosis and bilateral mastoiditis. The clinical findings of all affected family members were in agreement with the clinical diagnosis of OFCS and are summarized in Table 1.

A consanguineous family from Turkey with autosomal recessive otofaciocervical syndrome. a Pedigree of the family showing the homozygosity for markers on chromosome 20p11.22 in the affected individuals IV-9, IV-10, V-2, and V-3 with unusual and critical recombination breakpoints in affected individuals IV-10 and V-3. b Clinical photographs of the affected individual V-2 showing facial features, protruding shoulders, scapular winging and cutaneous syndactyly between the second and the third toe. c Affected individuals V-2, IV-9, and V-3 display typical facial features including cup-shaped ears, commissural lip clefting, and retrognathia
As the identification of the causative mutation out of hundreds of potential candidate variations in whole-exome sequencing (WES) data remains difficult and time consuming, we tested a strategy of pooling the DNAs of affected individuals before sequencing. This strategy allowed a fourfold reduction in costs and computing time compared to sequencing each affected family member individually. By performing WES on the single mixed sample of equal amounts of DNA of the affected individuals IV-9, IV-10, V-2, and V-3, a total of 45,609 variants were detected. In order to determine the effectiveness of our pooling strategy in reducing the number of homozygous variants, we compared the variant data set of the mixed OFCS sample with 11 data sets from WES of other patients, each from one index patient from a different consanguineous pedigree. As expected, the pooling of DNAs shifted the distribution of variants from high percentage of alternate reads (indicating homozygous presence of the variant) to lower percentages (Fig. 2a). When compared to the non-pooled samples, the mixed sample showed a twofold reduction in the number of variations in the 90–100 % range of alternate reads. To assess the reliability of the pooling strategy in determining homozygosity or heterozygosity for variants, we confirmed ten randomly selected variations from the WES data with a percentage of alternate reads between 75 and 80 % by Sanger sequencing of each affected individual. In all cases at least one individual was not homozygous for the variation and considering all alleles together, exactly 75 % of the alleles (60 of 80) showed the respective variation (Table 2).

Pooling DNA is a powerful strategy for the detection of homozygous variants in WES data from consanguineous families. a Distribution of variations in the pooled sample compared to non-pooled samples from consanguineous families. The y-axis shows the number of variations, expressed as percentage of the total variations. The x-axis shows the percentage of alternate alleles. Homozygous variations form the peak at the right end of the axis, while heterozygous variations form the middle peak. The distribution of variations in the pooled sample (light gray) shows a shift toward lower percentages of alternate alleles, when compared to the non-pooled samples (dark gray, n = 11). Error bars depict standard deviation. b After stringent filtering, the pooled sample (light gray) shows a striking decrease in the number of homozygous variations, when compared to other WES samples (dark gray, n = 11). Possibly homozygous variations (percentage of alternate alleles ≥75 %) are 4-fold decreased and very likely homozygous variations (percentage of alternate alleles ≥90 %) are 28-fold decreased. Error bars depict standard deviation
In a next step, we filtered variants according to their position relative to the exons, predicted effects, and frequency in the general population, as found in dbSNP, the 1000 genomes project, the exome variant server, and our in-house database (Table 3). After filtering, the WES data sets of the 11 controls had an average of 28 variants with a percentage of alternate reads ≥90 % and a coverage of at least five reads, while the mixed sample had only one, the c.497G>T (p.G166V) variant in PAX1 (Fig. 2b). We confirmed the presence and co-segregation of the variant with the disease in the family by Sanger sequencing of available DNAs from 11 family members (III-3, III-4, III-8, III-10, IV-5, IV-6, IV-9, IV-10, IV-11, V-2, and V-3; Fig. 1a). All four affected individuals were homozygous for this variant, whereas parents were found to be heterozygous carriers (Fig. 3a). The PAX1 c.497G>T variant is neither listed in 13,006 alleles from the exome variant server (http://evs.gs.washington.edu/EVS/) nor described as SNP in various other databases. Subsequent microsatellite marker analysis in the family further confirmed the homozygous PAX1 locus (Fig. 1a). Interestingly, individuals IV-10 and V-3 showed unusual and critical recombination breakpoints. Haplotyping defined a shared homozygous interval of only 1.5 Mb between markers D20S54 and D20S477 including PAX1 (Fig. 1a).

Identification of the c.497G>T mutation in PAX1. a Identification of the homozygous c.497G>T (p.G166V) PAX1 variant in patient V-2 (upper panel). The parents are heterozygous for this variant, shown for individual IV-6 (middle panel). Individual III-3 is shown as a control without PAX1 mutation (lower panel). b Schematic representation of described PAX1 transcripts in human (isoform 1, NM_006192.4; isoform 2, NM_001257096) and of the mouse ortholog (NM_008780.2), encoded amino acids (aa) of the PAX1 protein in human (NP_006183.2, NP_001244025.1) and mouse (NP_032806.2), and location of the identified mutation in human. c Analysis of Pax1 expression by RT-PCR in P6 mouse cochlea (control, water control). d Schematic representation of the PAX1 protein and conservation of the paired-box domain (aa 98-222, Pfam, and SMART database) of PAX1 containing the highly conserved glycine residue at position 166 in human and 157 in mouse (green paired box, blue octapeptide). The multiple sequence alignment was generated with ClustalW
The identified PAX1 p.G166V mutation is predicted to be probably damaging by PolyPhen-2 (score 1.000) and disease causing by MutationTaster (score 0.999), and it affects a highly conserved glycine residue (ConSeq score −0.964) within the DNA-binding paired-box domain. Therefore, we evaluated the effect of the mutation on the DNA-binding affinity of the mouse Pax1 ortholog (89 % identical and 91 % similar to human PAX1 isoform 1) to the regulatory sequence of the Nkx3-2 promoter region, a direct target of Pax1 transcriptional regulation, using a dual luciferase reporter assay in HEK293T cells (Fig. 4a). By co-transfection of the firefly reporter plasmid, the pRL-TK vector for normalization and either HA-tagged mouse Pax1 WT or Pax1 G157V (the glycine residue at position 157 in the mouse Pax1 protein corresponds to the glycine at position 166 in human PAX1), we observed a significantly reduced transactivation of the regulatory sequence of Nkx3-2 in cells overexpressing Pax1G157V compared to cells overexpressing Pax1WT (Fig. 4a). Subsequent Western blot analyses revealed comparable steady-state expression levels of Pax1WT and Pax1G157V proteins in HEK293T cells overexpressing Pax1 (Fig. 4b).

Reduced transactivation activity of Pax1G157V. a Relative luciferase activity driven by a regulatory sequence in the Nkx3-2 promoter region in HEK293T cells overexpressing Pax1G157V in comparison to Pax1WT expressing cells. Cells were cotransfected with expression vectors containing HA-tagged Pax1 WT or Pax1 G157V, the firefly reporter plasmid Nkx3-2-pGL4.10 and the pRL-TK vector for normalization. Control: Pax1 negative transfection with LacZ-pcDNA3.1. Three experiments were performed in triplicate. Error bars depict standard deviation. b Pax1 overexpression in HEK293T cells. Comparable steady-state expression levels of Pax1WT and mutant Pax1G157V proteins are shown by immunoblotting of whole cell lysates after measurement of the luciferase activity. Control: Pax1 negative transfection with LacZ-pcDNA3.1
Discussion
By performing WES on a single pooled sample of equal amounts of DNA of the four affected individuals, we were able to identify the c.497G>T (p.G166V) mutation in PAX1 in a consanguineous family from Turkey with autosomal recessive OFCS. PAX1 encodes a transcription factor expressed during the development of the skeleton, thymus, and parathyroid glands and required for normal development of ventral vertebral structures (Schnittger et al. 1992; Wallin et al. 1994, 1996). There are two PAX1 isoforms, differing in their length: The five exons of the large isoform 1 are transcribed into a 5,354-bp transcript (NM_006192.4), which encodes a protein of 534 amino acids (NP_006183.2), whereas the four exons of the isoform 2 are transcribed into a 5,364-bp transcript (NM_001257096) encoding a protein of 433 amino acids (NP_001244025.1; Fig. 3b). Like all proteins of the PAX gene family, PAX1 also contains a paired-box domain and an octapeptide (Fig. 3d). The identified mutation is located within the paired box of PAX1, a highly conserved sequence motif encoding the DNA-binding paired domain of transcription factors (Fig. 3d; Treisman et al. 1991; Chalepakis et al. 1991), which was first defined in different gene products in Drosophila (Bopp et al. 1986; Cote et al. 1987; Bopp et al. 1989). Subsequently, it has also been detected in other organisms, such as mouse and human (Deutsch et al. 1988; Burri et al. 1989). Using a dual luciferase reporter assay, we observed a significantly reduced transactivation of the regulatory sequence of the Nkx3-2 promoter region, a direct target of Pax1 transcriptional regulation in HEK293T cells overexpressing Pax1G157V compared to Pax1WT expressing cells. These results indicate a reduced DNA-binding affinity of the mutant protein Pax1G157V and demonstrate that the glycine residue at position 157 of Pax1 and position 166 in human PAX1 is critical for the binding of the transcription factor to the promoter region of target genes and that the p.G166V mutation represents a hypofunctional allele.
Previously, it was hypothesized that PAX1 variants might play a role in congenital vertebral malformation disorders including Klippel–Feil syndrome and Jarcho-Levin syndrome (Bannykh et al. 2003; McGaughran et al. 2003). Also, the identification of a PAX1 mutation in a patient with spina bifida suggests that PAX1 may act as a risk factor for human neural tube defects (Hol et al. 1996). Vertebral anomalies have also been observed in an undulated Pax1 mouse model showing decreased DNA-binding affinity and transcriptional activity of the mutant protein (Balling 1994; Chalepakis et al. 1991). However, Pax1 homozygous knockout mice demonstrated a phenotype with abnormalities of the tail, abnormal vertebral column, sternum, and scapula (Wilm et al. 1998). Our expression analysis of Pax1 by RT-PCR revealed expression in P6 mouse cochlea, which supports a role of Pax1 in the hearing process and is consistent with the hearing difficulties in affected individuals with PAX1 mutation c.497G>T (Fig. 3c). The underlying molecular pathogenesis of the identified p.G166V mutation is based on a reduced DNA-binding affinity of the Pax1G157V transcription factor likely leading to a decreased regulatory activity of PAX1 and reduced expression of PAX1 target genes including Nkx3-2 (Rodrigo et al. 2003). Notably, Nkx3-2 expression alterations have also been associated with oculoauriculovertebral spectrum (OAVS, MIM 164210) disorders (Fischer et al. 2006) causing skeletal anomalies and functionally linking OAVS disorders to OFCS.
Considering the phenotypic overlap of OFCS with other branchial-arch spectrum disorders, accurate discrimination of present symptoms is crucial for the correct diagnosis of affected individuals. In general, BORS, BOFS, and OFCS are characterized by microretrognathia, cleft lip/palate, alacrima, nasolacrimal duct stenosis, cup-shaped and low-set ears with preauricular fistulas, hearing loss, dental caries, and branchial defects. The presence of developmental delay and mild intellectual disability is mainly associated with BOFS or OFCS. In contrast to OFCS and BORS, patients with BOFS present with additional eye anomalies such as microphthalmia or anophthalmia, orbital dermoid cyst, and iris or retinal coloboma. Conjunctivitis can be found in patients with BORS or BOFS. The most important distinctive features of OFCS are the skeletal anomalies, which include vertebral defects, low-set clavicles, winged scapulae, and sloping shoulders, as well as the absence of renal involvement (Table 1). As mutations in EYA1 are described in both OFCS and BORS, it remains controversial whether the two syndromes are distinct entities or variable expression of the same disorder (Estefania et al. 2005; Dallapiccola and Mingarelli 1995; Rickard et al. 2001). PAX1 mutations might be restricted to OFCS patients, separating OFCS from other branchial-arch spectrum disorders. Nevertheless, it remains to be elucidated in future studies whether mutations in PAX1 are a more frequent cause of OFCS and are also found in patients with the clinical diagnosis of BORS as well.
We explored a WES strategy based on the pooling of the DNAs of all affected family members for identification of the causative variants. Of course, individual sequencing of the four affected individuals in our family would have led to the same result, but would have been more expensive and time consuming regarding the computational analysis. We compared the mixed OFCS sample with 11 data sets, each from one member of a different consanguineous pedigree. Before filtering, we observed a twofold reduction in the total number of homozygous variations. For a variant to be homozygous in the pooled sample, four different individuals from two branches of the family need to be homozygous. This is extremely unlikely for rare variants. Accordingly, after filtering out common variations, only very few homozygous variations should remain, including the one variant that was selected for the causative variant. In our analysis, we observed a 28-fold reduction in homozygous variations and were left with only one valid mutation, the c.497G>T mutation in PAX1. Moreover, we re-sequenced ten random variations from the mixed OFCS WES data set with a percentage of alternate reads between 75 and 80 %. In this range, we expect six of the eight pooled alleles to show the variation and two to be wild type. In all cases re-sequenced, 5–7 of the alleles showed the variation, with an average of exactly 6 (Table 2). Thus, we show that the DNA pooling strategy represents a powerful, straightforward, and cost-effective method for identifying causative variants in consanguineous pedigrees with several affected individuals. Interestingly, by confirming the PAX1 locus via microsatellite marker analysis, we observed unusual and critical recombination breakpoints in two affected individuals of the family. These recombination events would have made classical linkage analysis in this pedigree likely unsuccessful and demonstrate another advantage of the pooling strategy in WES. A limitation of the pooling strategy, however, is that it does not allow the investigation of alternative modes of inheritance. As such, the validity of the assumption that the causative mutation is homozygous by descent in all affected family members is critical for a successful analysis.
In conclusion, we identified PAX1 as a novel disease-causing gene for otofaciocervical syndrome and show that the pooling of DNA from affected individuals can reduce the number of putative disease-causing homozygous variants in consanguineous families, therefore representing a cost-effective and promising alternative to classical WES on ‘single’ DNA samples.
References
Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, Levi-Acobas F, Cruaud C, Le Merrer M, Mathieu M, Konig R, Vigneron J, Weissenbach J, Petit C, Weil D (1997a) Clustering of mutations responsible for branchio-oto-renal (BOR) syndrome in the eyes absent homologous region (eyaHR) of EYA1. Hum Mol Genet 6:2247–2255
Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, Weil D, Cruaud C, Sahly I, Leibovici M, Bitner-Glindzicz M, Francis M, Lacombe D, Vigneron J, Charachon R, Boven K, Bedbeder P, Van Regemorter N, Weissenbach J, Petit C (1997b) A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet 15:157–164
Balling R (1994) The undulated mouse and the development of the vertebral column. Is there a human PAX-1 homologue? Clin Dysmorphol 3:185–191
Bannykh SI, Emery SC, Gerber JK, Jones KL, Benirschke K, Masliah E (2003) Aberrant Pax1 and Pax9 expression in Jarcho–Levin syndrome: report of two Caucasian siblings and literature review. Am J Med Genet A 120A:241–246
Bopp D, Burri M, Baumgartner S, Frigerio G, Noll M (1986) Conservation of a large protein domain in the segmentation gene paired and in functionally related genes of Drosophila. Cell 47:1033–1040
Bopp D, Jamet E, Baumgartner S, Burri M, Noll M (1989) Isolation of two tissue-specific Drosophila paired box genes, Pox meso and Pox neuro. EMBO J 8:3447–3457
Burri M, Tromvoukis Y, Bopp D, Frigerio G, Noll M (1989) Conservation of the paired domain in metazoans and its structure in three isolated human genes. EMBO J 8:1183–1190
Chalepakis G, Fritsch R, Fickenscher H, Deutsch U, Goulding M, Gruss P (1991) The molecular basis of the undulated/Pax-1 mutation. Cell 66:873–884
Chi N, Epstein JA (2002) Getting your Pax straight: Pax proteins in development and disease. Trends Genet 18:41–47
Cote S, Preiss A, Haller J, Schuh R, Kienlin A, Seifert E, Jackle H (1987) The gooseberry-zipper region of Drosophila: five genes encode different spatially restricted transcripts in the embryo. EMBO J 6:2793–2801
Dallapiccola B, Mingarelli R (1995) Otofaciocervical syndrome: a sporadic patient supports splitting from the branchio-oto-renal syndrome. J Med Genet 32:816–818
Deutsch U, Dressler GR, Gruss P (1988) Pax 1, a member of a paired box homologous murine gene family, is expressed in segmented structures during development. Cell 53:617–625
Estefania E, Ramirez-Camacho R, Gomar M, Trinidad A, Arellano B, Garcia-Berrocal JR, Verdaguer JM, Vilches C (2005) Point mutation of an EYA1-gene splice site in a patient with oto-facio-cervical syndrome. Ann Hum Genet 70:140–144
Fara M, Chlupackova V, Hrivnakova J (1967) Familial oto-facio-cervical dysmorphia. Acta Chir Orthop Traumatol Cech 34:511–520
Fischer S, Ludecke HJ, Wieczorek D, Bohringer S, Gillessen-Kaesbach G, Horsthemke B (2006) Histone acetylation dependent allelic expression imbalance of BAPX1 in patients with the oculo-auriculo-vertebral spectrum. Hum Mol Genet 15:581–587
Hol FA, Geurds MP, Chatkupt S, Shugart YY, Balling R, Schrander-Stumpel CT, Johnson WG, Hamel BC, Mariman EC (1996) PAX genes and human neural tube defects: an amino acid substitution in PAX1 in a patient with spina bifida. J Med Genet 33:655–660
Kaiser R, Posteguillo EG, Muller D, Just W (2007) Exclusion of genes from the EYA-DACH-SIX-PAX pathway as candidates for Branchio-Oculo-Facial syndrome (BOFS). Am J Med Genet A 143A:2185–2188
Lang D, Powell SK, Plummer RS, Young KP, Ruggeri BA (2007) PAX genes: roles in development, pathophysiology, and cancer. Biochem Pharmacol 73:1–14
McGaughran JM, Oates A, Donnai D, Read AP, Tassabehji M (2003) Mutations in PAX1 may be associated with Klippel-Feil syndrome. Eur J Hum Genet 11:468–474
Noll M (1993) Evolution and role of Pax genes. Curr Opin Genet Dev 3:595–605
Read AP, van Heyningen V (1994) PAX genes in human developmental anomalies. Semin Dev Biol 5:323–332
Rickard S, Boxer M, Trompeter R, Bitner-Glindzicz M (2000) Importance of clinical evaluation and molecular testing in branchio-oto-renal (BOR) syndrome and overlapping phenotypes. J Med Genet 37:623–627
Rickard S, Parker M, van‘t Hoff W, Barnicoat A, Russell-Eggitt I, Winter RM, Bitner-Glindzicz M (2001) Oto-facio-cervical (OFC) syndrome is a contiguous gene deletion syndrome involving EYA1: molecular analysis confirms allelism with BOR syndrome and further narrows the Duane syndrome critical region to 1 cM. Hum Genet 108:398–403
Rodrigo I, Hill RE, Balling R, Munsterberg A, Imai K (2003) Pax1 and Pax9 activate Bapx1 to induce chondrogenic differentiation in the sclerotome. Development 130:473–482
Ruf RG, Xu PX, Silvius D, Otto EA, Beekmann F, Muerb UT, Kumar S, Neuhaus TJ, Kemper MJ, Raymond RM Jr, Brophy PD, Berkman J, Gattas M, Hyland V, Ruf EM, Schwartz C, Chang EH, Smith RJ, Stratakis CA, Weil D, Petit C, Hildebrandt F (2004) SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc Natl Acad Sci USA 101:8090–8095
Schnittger S, Rao VV, Deutsch U, Gruss P, Balling R, Hansmann I (1992) Pax1, a member of the paired box-containing class of developmental control genes, is mapped to human chromosome 20p11.2 by in situ hybridization (ISH and FISH). Genomics 14:740–744
Treisman J, Harris E, Desplan C (1991) The paired box encodes a second DNA-binding domain in the paired homeo domain protein. Genes Dev 5:594–604
Wallin J, Wilting J, Koseki H, Fritsch R, Christ B, Balling R (1994) The role of Pax-1 in axial skeleton development. Development 120:1109–1121
Wallin J, Eibel H, Neubuser A, Wilting J, Koseki H, Balling R (1996) Pax1 is expressed during development of the thymus epithelium and is required for normal T-cell maturation. Development 122:23–30
Wilm B, Dahl E, Peters H, Balling R, Imai K (1998) Targeted disruption of Pax1 defines its null phenotype and proves haploinsufficiency. Proc Natl Acad Sci USA 95:8692–8697
Acknowledgments
We are grateful to all family members who participated in this study, to Esther Milz for excellent technical assistance, to Andrea Pannes for her technical advice in slow-down PCR, to Simon von Ameln for cochlea preparation, and to Karin Boss for critically reading the manuscript. This work was supported by the German Federal Ministry of Education and Research (BMBF) by grant number 01GM1211A (E-RARE network CRANIRARE-2) to B.W.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pohl, E., Aykut, A., Beleggia, F. et al. A hypofunctional PAX1 mutation causes autosomal recessively inherited otofaciocervical syndrome. Hum Genet 132, 1311–1320 (2013). https://doi.org/10.1007/s00439-013-1337-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-013-1337-9