
Abstract
RNA sequencing analysis was carried out to characterize egg and larval transcriptomes in the appendicularian, Oikopleura dioica, a planktonic chordate, which is characterized by rapid development and short life cycle of 5 days, using a Japanese population of the organism. De novo transcriptome assembly matched with 16,423 proteins corresponding to 95.4 % of the protein-encoding genes deposited in the OikoBase, the genome database of the Norwegian population. Nucleotide and amino acid sequence identities between the Japanese and Norwegian O. dioica were estimated to be around 91.0 and 94.8 %, respectively. We discovered 175 novel protein-encoding genes: 144 unigenes were common to both the Japanese and Norwegian populations, whereas 31 unigenes were not found in the OikoBase genome reference. Among the total 12,311 unigenes, approximately 63 % were detected in egg-stage RNAs, whereas 99 % were detected in larval stage RNAs; 3772 genes were up-regulated, and 1336 genes were down-regulated more than four-fold in the larvae. Gene ontology analyses characterized gene activities in these two developmental stages. We found a messenger RNA (mRNA) 5′ trans-spliced leader, which was observed in 40.8 % of the total unique transcripts. It showed preferential linkage to adenine at the 5′ ends of the downstream exons. Trans-splicing was observed more frequently in egg mRNAs compared with larva-specific mRNAs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The appendicularian, Oikopleura dioica, is a marine planktonic tunicate that retains a swimming tadpole shape through its entire life. This animal possesses a number of advantages as a model organism (Nishida 2008): (1) It has a short life cycle (about 5 days at 20 °C); (2) Its development is rapid, and organogenesis is complete within 10 h after fertilization to form a functional body (Fig. 1); (3) Its morphogenesis and cell linages are well described (Fujii et al. 2008; Nishida 2008; Nishida and Stach 2014; Stach et al. 2008); (4) Live imaging of embryos by introducing fluorescent protein messenger RNAs (mRNAs) is feasible (Kishi et al. 2014); (5) The RNAi method is available for knockdown of zygotic mRNA as well as maternal mRNAs in the ovary and eggs (Omotezako et al. 2013); and (6) It has a compact and fully sequenced genome of 70 Mb, the smallest ever found in a chordate (Denoeud et al. 2010; Seo et al. 2001). The number of genes is estimated to be approximately 18,000, indicating a high gene density (one gene per 5 kb in the genome) (Denoeud et al. 2010; Seo et al. 2001). These features make O. dioica a useful organism for studies of development and genome plasticity in a tunicate with a short generation time (Delsuc et al. 2006).

Oikopleura dioica. a Unfertilized eggs. b Late larvae at 8 hpf. c Functional juveniles at 10 hpf after the tail shift. Developmental times at 20 °C are shown. Specimens were collected at the stages of (a) and (b). Asterisk indicates the position of the mouth. Scale bar = 100 μm
Genome-wide knowledge of the transcriptome provides a resource for understanding gene functions underlying the formation of a functional body. In O. dioica, tiled microarray analysis using genomic DNA probes has been carried out (Danks et al. 2013). The genome browser, OikoBase, showed that 77 % of predicted genes (13,081 out of 16,749 tested genes) are expressed at some point from embryogenesis through larval morphogenesis (Danks et al. 2013). The OikoBase includes expressed sequence tags (ESTs) and microarray data. Deep RNA sequencing (RNA-Seq) is expected to provide more information, such as sequence polymorphism and novel genes. Moreover, little is known about intra-species genetic diversity. In the case of ascidians, e.g., Ciona intestinalis and Ciona savignyi, a group of tunicates, there are high levels of intra-species nucleotide polymorphism and amino acid substitution among geographically distant populations in each species (more than 10 times the level found in vertebrates) (Caputi et al. 2007; Griggio et al. 2014; Nydam and Harrison 2010; Suzuki et al. 2005). An O. dioica sequence data set has been available for the Norwegian population (Denoeud et al. 2010; Seo et al. 2001). In-depth RNA sequencing of the Japanese O. dioica would not only yield a resource of maternal and zygotic transcriptomes but also provide an opportunity for intra-species comparison of whole exon sequences, i.e., the exome, to gain insight into genome plasticity in this rapidly evolving metazoan.
In the present study, we carried out an RNA-Seq analysis of a Japanese population of O. dioica. In order to obtain maternal and zygotic transcript sequences, we used the whole organism at two developmental stages: the unfertilized egg and the larva during organogenesis. De novo assembly (Grabherr et al. 2011) of reads recovered 16,423 proteins corresponding to 95.4 % of protein-encoding genes that were predicted in the genome of the Norwegian O. dioica. Furthermore, the depth of sequence data contributed to identification of 175 novel protein-encoding genes. Transcriptome-wide comparison revealed high levels of sequence variation between the two O. dioica populations. Gene ontology (GO) analysis characterized the features of gene activities at these two developmental stages. A 5′ trans-spliced leader (SL), as previously reported, was also found (Denoeud et al. 2010; Ganot et al. 2004).
Materials and methods
Laboratory culture and sample collection
Live wild specimens were collected at Sakoshi Bay and Tossaki port in Hyogo, Japan, by scooping surface seawater with a bucket. O. dioica were sorted and cultured in the laboratory over generations as described previously (Bouquet et al. 2009; Omotezako et al. 2013). In brief, they were reared in 10-l containers in artificial seawater (REI-SEA Marine, REI-SEA, Tokyo, Japan) stirred with a paddle (15 rpm) at 20 °C and fed the flagellates Isochrysis galbana and Rhinomonas reticulata, the diatom Chaetoceros calcitrans, and the cyanobacterium Synechococcus sp. O. dioica becomes sexually mature and spawns at 5 days post-fertilization.
Unfertilized eggs (Fig. 1a) and larvae at 8 h post-fertilization (hpf) (Fig. 1b) were used in this study. To minimize inter-individual allelic variations in the sequencing results, samples were prepared from cohorts of a single pair. Therefore, all sequence data are derived from cohorts of the same pair. A male and a female were transferred to a 2-l container to allow spawning, and the cohorts were reared over several generations. Unfertilized eggs were obtained from 46 female cohorts. Larval samples were collected from cohorts of 63 adult pairs. Matured females were placed in gelatin-coated 6-well culture plates to allow natural spawning. Oocytes were collected in a petri dish and fertilized by adding drops of seawater containing sperm. After three times of washing to remove sperm, they were cultured at 20 °C. In this condition, animals complete organogenesis and become functional juveniles in 10 h (Fig. 1c). Larvae at 8 hpf were anesthetized and collected in 1.5-ml tubes. Eggs and larvae were frozen immediately in liquid nitrogen and stored at −80 °C until isolation of total RNA.
RNA isolation, library construction, and RNA-Seq
Total RNA was isolated by guanidium thiocyanate-phenol-chloroform extraction. The OD260/280 and OD260/230 were 2.09–2.31 and 1.88–1.92, respectively, ensuring purity of the RNA samples. The amount of total RNA in each sample was more than 20 g. Integrity of total RNA was confirmed using agarose gel electrophoresis and an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA, USA). Illumina TruSeq RNA Preparation Kit (Illumina, Japan) was used for library construction. Strand-specific RNA-Seq was performed at Beijing Genome Institute (BGI, Shenzhen, China) using a paired-end library. In brief, mRNA was purified with oligo deoxythymine (dT) and fragmented. The first-strand complementary DNA (cDNA) was synthesized using random hexamers, and a strand-oriented library was generated. Libraries for the oocyte and larva were sequenced using Illumina HiSeq™ 2000. The insert size of the library and the read length was 200 and 90 bp, respectively. The raw data were deposited in the National Center for Biotechnology Information (NCBI) Short Read Archive (SRP accession number: SRP050571, run accession numbers are SRR1693762, SRR1693765, SRR1693766, and SRR1693767) and Gene Expression Omnibus (GEO accession: GSE64421).
Data filtering and de novo transcriptome assembly
In order to achieve high-quality transcriptome assembly, all raw reads were filtered using the following steps: (1) Adapter and contamination sequences were removed using FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/). (2) Low-quality bases whose quality was less than 25 from both the 5′ and 3′ ends were trimmed. (3) Sequences with a length of less than 35 nt were discarded. (4) Sequences with Q20 or unknown (Ns) bases accounting for more than 10 % were also discarded. (5) Unpaired reads after the above processes were removed.
Reads of the egg and larval stages were merged, and de novo transcriptome assembly was carried out using SOAPdenovo-Trans and Trinity assemblers. These are the most frequently used tools for transcriptome assembly. We set the k-mer parameter as 25 for both assemblers.
Identification of known and novel protein-encoding genes
To identify known potential protein-encoding genes, all assembled transcripts were queried against OikoBase proteins and the UniProtKB/Swiss-Prot and NCBI RefSeq databases using BLASTX (E value cutoff at 1E-5). Best-hit transcripts for each protein in these databases were retained as “known unigenes”. “Novel unigenes” were identified among remaining transcripts using a local pipeline, which is summarized in Fig. 3a. Specifically, sequences that corresponded to isoforms of the known unigenes were first removed. Next, potential non-coding RNAs were detected and excluded with a cmsearch (Nawrocki and Eddy 2013) E value of ≥0.01 against the Rfam database (Burge et al. 2013), and only transcripts with a length of ≥500 bp were retained. Finally, three frames of these transcripts were obtained by transeq in the EMBOSS (Rice et al. 2000) package, and hmmsearch (Eddy 2011) was used to identify potential Pfam domains (E value of ≤0.001). The corresponding transcripts with at least one Pfam domain were considered to be novel protein-encoding genes. Only the longest isoforms were kept in the novel unigene set.
RT-PCR analysis
Expression of six known unigenes and 18 novel unigenes was validated by reverse transcriptase PCR (RT-PCR). Total RNA was extracted from eggs and 8 hpf larvae using TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA). Total RNA (500 ng) was reverse-transcribed into single-stranded cDNA using an oligo dT primer and Superscript III Reverse Transcriptase (Invitrogen). RT-PCR reactions were performed with 0.1 μM primer and 5 ng cDNA using TaqDNA polymerase (New England Biolabs, Ipswich, MA, USA) or KOD-Plus (Toyobo, Osaka, Japan). The PCR was carried out at 94 °C for 2 min, followed by 35 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 1 min. The primers (Supplementary Table S1) were designed on the basis of the assembled transcript sequences. The electrophoresis images were captured using a FAS-IV gel imaging system (NIPPON Genetics, Tokyo, Japan).
Quantitative analysis of gene expression via RNA-Seq read abundance
To characterize maternal and zygotic genes, quantitative analysis of read numbers in eggs and late larvae was performed. The amounts of mRNAs were calibrated by the fragments per kilobase of exon per million fragments mapped (FPKM) method (Li and Dewey 2011). For quantification of fold changes (FC) in reads between larvae and egg, 3 was added to the expression value in order to exclude rarely expressed genes, following a similar protocol of Adiconis et al. (Adiconis et al. 2013). Genes with a FPKM that changed at least 4-fold were regarded as up-regulated or down-regulated genes.
GO annotation and enrichment analysis
Genes that were differentially expressed between egg and larva were again queried against UniProtKB/Swiss-Prot (E value of 1.0E-5) using BLASTX. The corresponding symbols for each unigene were retrieved according to the best hit and then submitted to Ontologizer (Bauer et al. 2008) for functional enrichment analysis. Currently, the O. dioica gene association file is not available. Thus, human genes, which had the most gene entries matched, were used as the background for GO enrichment analysis. “Parent-child-union” method was used, and p value was adjusted via Bonferroni correction.
Identification of 5′ SL sequences and trans-spliced mRNA
Unigenes were queried against themselves using megablast with a word size of 15 and an E value threshold of 1000, using the maximum number of aligned sequences possible. Only forward vs forward matches were considered, and self vs self matches were excluded. Matched positions of query and subject sequences had to be between the 1st and 150th base pairs of the transcript, and at least one of the aligned sequences had to start from 1 at the 5′ end. The aligned sequences were extracted and clustered using CD-HIT-EST (Fu et al. 2012), and multiple sequence alignment was performed using MUSCLE (Edgar 2004). SLs were identified by manually checking the conserved motif in the multiple sequence alignment. mRNAs that had the SL sequence at the 5′ end were regarded as trans-spliced mRNAs. Additional mRNAs with partial stretches, but more than 10 bp, of the identified spliced leaders were also regarded as trans-spliced mRNAs.
Results
De novo transcriptome assembly
To obtain information on maternal and zygotic transcripts, RNA-Seq was carried out using poly(A) + RNAs collected from unfertilized eggs (Fig. 1a) and late larvae at 8 hpf (Fig. 1b). After data filtering, approximately 97 million clean reads (∼8.7 Gbp) with an average length of 89 bp were obtained (48,712,280 and 48,365,226 reads from egg and larva, respectively) (Supplementary Table S2). We tried to map the RNA-Seq reads to the genome reference stored in the OikoBase (Danks et al. 2013). However, only about 10 % of reads could be mapped, and the result was not greatly improved even if we used a much looser parameter. Moreover, genome-guided Trinity (Haas et al. 2013) assembly generated a mapping rate of only 69.9 % with N50 as 371 bp (data not shown). The genome sequence in the OikoBase is derived from O. dioica collected in Norway. Here, we analyzed specimens that were collected in Japan. Our preliminary sequencing analyses of some cDNAs, such as brachyury, muscle actin 3, and testis-specific histones, demonstrated sequence variation between these geographically distant O. dioica populations (data not shown). The low mapping efficiency might have been due to intra-species sequence differences. In the present study, therefore, we chose de novo transcriptome assembly.
The results of SOAPdenovo-Trans (Xie et al. 2014) and Trinity (Haas et al. 2013) de novo transcriptome assembly are shown in Supplementary Table S2. SOAPdenovo-Trans generated a total of 72,996 scaffolds with a length greater than 100 bp, and the total length of these scaffolds was ∼36.16 Mbp. The length distribution of the scaffolds is shown in Fig. 2a. Among the reads, 77.1 % were mapped to the assembled transcriptome using Tophat2 (version 2.0.11) (Kim et al. 2013), and 65.0 % were concordant pairs. By contrast, Trinity generated a much larger transcriptome, with a total of 86,898 transcripts account for ∼70.80 Mbp. N25, N50, and N75 were much larger than was indicated by SOAPdenovo-Trans. Moreover, the mapping rate of 89.2 % of total reads and the mapped pair concordance rate of 81.4 % were much better than for SOAPdenovo-Trans. CD-HIT-EST (version 4.6.1) (Li and Godzik 2006) redundancy analysis at 90 % threshold showed that 68.5 % of the Trinity transcriptome was non-redundant, being much less than the value of 89.1 % for the SOAPdenovo-Trans transcriptome. This was due to the difference in scaffolding strategies between SOAPdenovo-Trans and Trinity. Unlike SOAPdenovo-Trans, Trinity does not join gapped contigs with Ns but reports alternative splicing forms as different transcripts. This is why the total length of the Trinity transcriptome is greater than that of SOAPdenovo-Trans. Considering all these factors, we considered Trinity to be a better choice for de novo transcriptome assembly and used the results of Trinity for further analyses in this study.

De novo transcriptome assembly and analysis. a Length distribution of scaffolds assembled by SOAPdenovo-Trans and Trinity de novo assembly. b Length distribution of assembled transcripts with and without significant matches with known protein-encoding genes. c Sequence similarity between the Japanese and Norwegian populations at the nucleotide (nucl) and amino acid (prot) level
Recovery of most of the predicted genes in the O. dioica genome browser by annotation of assembled transcripts
Next, we annotated the assembled transcripts with known protein-encoding transcripts on the basis of homology to proteins in public databases (Table 1). Using a BLASTX E value cutoff at 1E-5, we identified a total of 55.2 % transcripts that had at least one hit against the three public databases shown in Table 1. In detail, 52.2 % of the transcripts matched OikoBase proteins (Danks et al. 2013), 29.1 % matched the UniProtKB/Swiss-Prot database (Boutet et al. 2007), and 31.4 % matched the NCBI RefSeq database (Pruitt et al. 2005); 44.8 % of the 86,898 transcripts did not show any significant matches with the aforementioned databases. These non-matching transcripts accounted for only 10 Mb of the transcriptome length of 71 Mb, indicating that these unmatched fragments are much shorter than the annotated transcripts. Indeed, these fragments correspond to the short transcript groups in the length distribution histogram (Fig. 2b). Most transcripts without any significant hit to known protein-encoding genes may be non-coding RNAs (such as ribosomal RNAs, tRNAs, lncRNAs), transcripted cis-regulatory regions (such as UTR and intron regions) of known protein-encoding genes, novel protein-encoding genes, or transcripts that have been misassembled (Wu et al. 2012).
To determine the percentage recovery of O. dioica genes, the assembled transcripts were aligned with sequence data set in the OikoBase, which predicted 17,212 protein products in the genome of the Norwegian O. dioica (Danks et al. 2013). Our analysis showed that 45,368 (52.2 %) of the assembled transcripts encoded proteins that have homology with 16,423 OikoBase proteins (Table 1). This means that 95.4 % of the predicted proteins in the OikoBase were thus recovered using an E value cutoff of 1.0E-5. Higher E value thresholds were also tested: 48.3 and 41.1 % of transcripts recovered 16,140 (93.8 %) and 15,582 (90.5 %) OikoBase proteins with E values of 1.0E-10 and 1.0E-20, respectively. Our analysis further showed that the assembled transcripts recovered most cDNA clones in the Norwegian O. dioica ESTs (103,969 clones in total) (Denoeud et al. 2010). Our transcripts showed homology with 97.4, 96.6, and 94.4 % of clones using E value thresholds of 1.0E-5, 1.0E-10, and 1.0E-20, respectively. These results suggest that our assembly of RNA-Seq reads could recover most of the gene transcripts predicted in the genome of the Norwegian O. dioica.
Trinity sequence assembly generated 57,962 non-redundant unigenes (according to the generated gene id by Trinity), which are about three times the number of predicted genes in OikoBase (Danks et al. 2013). In order to remove redundancy, only unigenes that showed the best hits with OikoBase proteins were screened. Accordingly, 12,136 genes corresponding to 16,423 (95.4 % of 17,212) OikoBase proteins were left (Supplementary Table S3). These were considered to be “known” unigenes. A single unigene would correspond to multiple duplicated genes, multiple paralogs, and possibly some polycistronic operons in OikoBase. In the O. dioica genome, 1761 operons containing 4997 genes have been predicted (Denoeud et al. 2010). All of the 12,136 known unigenes also showed homology with proteins in the UniProtKB/Swiss-Prot or NCBI RefSeq databases with an E value of 1.0E-5.
Novel protein-encoding genes
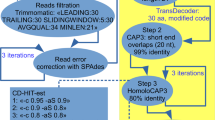
Next, we tested whether there were any remaining novel protein-encoding genes that had not been discovered in the aforementioned processes due to a lack of sequence homology or genome annotation. As shown in Fig. 3a, we screened the 38,935 (44.8 %) of transcripts that did not show any matches with known sequences. In brief, we removed isoforms of known genes (35,210 transcripts remained), excluded possible non-coding RNAs (35,017 transcripts remained), and eliminated transcripts that were shorter than 500 bp (2773 transcripts remained). As a result, 320 potential protein-encoding transcripts (including possible alternative splicing forms) belonging to 175 “novel” unigenes were obtained (Supplementary Table S3). None of them showed any homology with known proteins in public databases (OikoBase, UniProtKB/Swiss-Prot, and NCBI RefSeq databases) with an E value of ≤1.0E-5, but they possessed known Pfam domains as a part of the protein sequence (Fig. 3a). To ascertain whether these novel genes are expressed, 18 of them were randomly selected and RT-PCR assays were carried out. As shown in Fig. 3b, c, all of the transcripts were detected in eggs or larvae (8 hpf), suggesting the presence of uncharacterized unigenes in the Japanese O. dioica.

Identification of novel protein-encoding genes. a Flowchart of the pipeline for identification of novel protein-encoding genes. b, c RT-PCR validation of 18 novel genes with trans-splicing leaders in eggs or larvae (8 hpf) with and without reverse transcriptase (RT). Expression of the 11 unigenes whose sequences are present in the OikoBase Norwegian genome reference (b) and 7 unigenes that are absent in it (c). Arrows indicate the band with expected length of amplification. Extra bands are observed due to amplication of non-specific RT-PCR products
The genome annotation of the Norwegian O. dioica revealed 175 novel unigenes that had not been found previously (Denoeud et al. 2010). To determine whether these 175 novel unigenes are present in the Norwegian O. dioica genome, we queried all of them against the Norwegian genome reference. BLASTN (E value of ≤1E-30) revealed that 144 of the unigenes (including 11 PCR-confirmed genes in Fig. 3b) are in the Norwegian genome, and 108 of them have at least one intron in the Norwegian genome reference. In contrast, the other 31 novel unigenes (including 7 PCR-confirmed genes in Fig. 3c) were not found or had only a small part matched in the genome reference. These results demonstrated that the depth of RNA-Seq analysis contributes to identification of novel gene products that have been missed by traditional technology. Thus, a total of 12,311 (12,136 known plus 175 novel) unique protein-encoding genes were discovered (Supplementary Table S3). These were used for subsequent expression and functional analyses.
Transcriptome-wide sequence comparison between the Japanese and Norwegian O. dioica
To understand the degree of sequence conservation between the Japanese (our data) and Norwegian (OikoBase) populations, the 12,136 known unigenes were queried against the OikoBase transcript reference with BLASTN (word size of 15). BLAST hits with an alignment length of ≤300 (nucleotide level) or an E value of ≥1E-10 were discarded. Large gaps in the BLAST results were removed in nucleotide and protein levels. Genes with multiple hits, which may include paralogs and duplicated genes, were also excluded in order to avoid inaccuracy. As a result, 4843 and 7739 one-to-one hits were obtained at nucleotide and protein levels, respectively. A total of 4136 unigenes showed one-to-one hits at both the nucleotide and protein level and subjected to global sequence alignment. The sequence identity (from the first high-scoring segment pairs) was 91.0 and 94.8 % on average at the nucleotide and amino acid level, respectively. Of the 4136 sequences, 94.6 % showed 80 to 95 % identity at the nucleotide level, and 98.8 % of the sequences showed 80 to 100 % identity at the amino acid level (Fig. 2c, left panel). To ascertain effects of read coverage on the polymorphisms, we re-assessed the data using 1024 unigenes with higher coverage (FPKM ≥ 100) and 663 unigenes with lower coverage (FPKM ≤ 10) (Fig. 2c, middle and right panels). The sequence identity of unigenes with high coverage was 92.5 and 96.7 % on average at the nucleotide and amino acid level, respectively. Most unigenes in this group showed 90 to 100 % identities in both nucleotide and protein levels. In comparison, the sequence identity of unigenes with lower coverage was 89.1 and 92.2 % on average at the nucleotide and amino acid level, respectively. Taken together, the data indicated a possibility of marked intra-species sequence variation between the Japanese and Norwegian O. dioica.
Characterization of maternally expressed genes and zygotically expressed genes
We re-assembled the transcriptomes for the egg and larval stages separately. Querying the 12,311 identified unigenes against the assembled transcripts with BLASTN (E value of ≤1E-30), we found that approximately 63 % were detected in egg-stage RNAs whereas 99 % were detected in larval stage RNAs. In order to further evaluate maternal and zygotic transcripts, quantitation of read numbers in eggs and larvae was carried out. The amounts of transcripts for the 12,311 unigenes were calibrated using the fragments per kilobase of exon per million fragments mapped (FPKM) method (Li and Dewey 2011). To validate the quantification, we first tested three well-characterized genes: Brachyury, Muscle actin, and Vasa (Supplementary Table S4). Brachyury and Muscle actin are typical zygotic genes, and their expression becomes detectable in 32- to 64-cell embryos, respectively (Bassham and Postlethwait 2000; Nishida 2008), while Vasa is a maternally expressed gene involved in germ cell development, and its mRNA is detectable in both eggs and larvae (Shirae-Kurabayashi et al. 2006). Our comparison of read numbers was in agreement with these previous in situ hybridization studies. The read numbers of Brachyury and Muscle actin transcripts in larvae were over 30-fold more than those in eggs, while the number for the Vasa transcript at the two stages was almost the same.
Next, fold changes in read numbers from eggs to larvae were calibrated by adding 3 to the expression values in order to exclude artifacts in rarely expressed genes, using a modification of the method of Adiconis et al. (Adiconis et al. 2013). We found that 5108 genes showed a change of at least 4-fold (Supplementary Table S3): 3772 genes (74 %) were up-regulated from egg to larva, and 1336 genes (26 %) were down-regulated (Fig. 4a). Therefore, more than 40 % of unigenes could be categorized as up- or down-regulated genes. Because we did not prepare biological replicates in the present study, we carried out RT-PCR to ascertain expression of six known genes (Noa36, Mago, Tcf3, Par6, Spectrin, and Alpha3). As shown in Fig. 4b, Noa36 and Mago mRNA levels in eggs were higher than those in larvae. By contrast, Tcf3 mRNA level in larva was higher than that in eggs (Fig. 4b). Other three genes did not show any detectable differences between the two developmental stages. These expression patterns coincided well with the RNA-Seq results. The expression values are available in Supplementary Table S3.

Differentially expressed genes. a Scale plot of up-regulated and down-regulated genes. Three was added to every expression value (FPKM) and plotted on log2 axes. b RT-PCR validation of the expression of some genes during egg and larval stages. All of the expression patterns were coincident with our RNA-Seq result. Arrows indicate the band with expected length of amplification
Gene ontology (GO) (Harris et al. 2004) terms were assigned to the 3772 up-regulated and 1336 down-regulated genes. Enrichment analysis was performed to demonstrate conspicuous biological processes at each stage (Supplementary Table S5). Most of the up-regulated genes were involved in localization and transport processes (such as transmembrane transport, organic anion transport, single -organism transport, and localization), metabolic process (such as organonitrogen compound metabolism, sulfur compound metabolism, aromatic amino acid family metabolism, carbohydrate derivative metabolism), developmental process, organ morphogenesis, biological adhesion, synaptic transmission, cell junction assembly, and cell-cell signaling. By contrast, most down-regulated genes were involved in a cell cycle process, various forms of DNA or RNA processing (such as mRNA metabolism, establishment of RNA localization, DNA repair, DNA replication, DNA-templated transcription, elongation, and transcription elongation from RNA polymerase II promoter), and biogenesis (ribonucleoprotein complex biogenesis, cellular component organization, or biogenesis).
Pre-mRNA 5′ spliced leader
Spliced leader (SL) trans-splicing is a unique RNA splicing process in which a short spliced exon from one RNA transcripts links to the 5′ end of another RNA transcript. SL trans-splicing occurs broadly in tunicates (Ganot et al. 2004; Gasparini and Shimeld 2011; Matsumoto et al. 2010; Satou et al. 2006; Vandenberghe et al. 2001). In this study, we identified a total of 5020 trans-splicing mRNAs among the 12,311 unigenes, indicating that 40.8 % of mRNA species are trans-spliced, at least in some of those mRNAs.
These trans-spliced mRNAs are using only one SL, and the detected consensus sequence is AGUCCGAUUUCGAUUGUCUAACAG. While this sequence is homologous to the previously reported SL (Ganot et al. 2004), 16 bases at the 5′-end of the previously reported SL were not detected in our analysis. This may have happened because we used the Illumina TruSeq RNA Sample Preparation Kit, which could not completely capture 5′ end of mRNA. We then checked the pre-trimmed reads and found the perfect match of the whole previously reported SL, but in most case, some 5′ end bases were lost. To ascertain whether additional trans-splicing mRNAs were detectable, we queried the whole SL sequence including the previously reported sequence against the 12,311 unigenes, again using BLASTN with a smaller word size of 10, but no additional trans-splicing mRNA was obtained.
Next, we investigated whether the SL has any preferred bases by plotting the first three bases in the immediately linked downstream exons. As shown in Fig. 5a, it tended to link to exons having an A-enriched header. Adenine accounted for 86 and 52 % of the first and second bases, showing a specific preference for the first and second positions, while thymine accounted for the third base in 49 % of cases.

The preferential base of detected spliced leader and distribution of trans-spliced mRNAs in the egg, larvae. a Preference of the first three bases in the immediately linked downstream exons of SL. b Distribution of trans-spliced (SL) and non-trans-spliced (Non-SL) mRNAs in eggs or larvae (upper panels) and down-regulated genes or up-regulated genes (lower panels)
We examined whether the frequency of trans-splicing differs between eggs and larvae. SL was found in 4565 unigenes (58.5 % of unigenes found in egg-stage mRNAs) at the egg stage and 4469 unigenes (36.6 % unigenes found in larval stage mRNAs) at the larval stage (Fig. 5b). Thus, the number of mRNA species with this SL is almost the same, but the ratio of the trans-spliced mRNA species is reduced at the larval stage. Likewise, 1034 (20.6 %) of total trans-spliced mRNAs with the SL belong to the down-regulated genes (fold change ≤−4) while 422 (8.4 %) of them belong to the up-regulated genes (fold change ≥4) (Fig. 5b). Comparison of the unigenes with SL between the oocyte and larval stages showed that 3858 unigenes are commonly trans-spliced (matched to at least 12 bp from the 3′ end of the SL). Therefore, it is likely that SL is observed more frequently in maternal mRNAs in eggs when compared with zygotic mRNA in larvae.
To examine whether the trans-splicing prefers genes with special functions, the GO terms for trans-spliced unigenes and those of non-trans-spliced unigenes were compared (Supplementary Table S6). The genes with SL show similar GO enrichments with the case of the down-regulated genes (Supplementary Table S5). For instance, they included RNA processing processes and cell cycle-associated processes. On the other hand, the genes without detectable SL show similar enrichments with the case of the up-regulated genes (Supplementary Table S5), including localization, transport, developmental, and metabolic processes. The similar functional enrichments between the unigenes with SL and “down-regulated” genes suggest a possible involvement of trans-spliced mRNAs in early embryogenesis.
Discussion
In this study, we carried out an RNA-Seq analysis of a Japanese O. dioica population to gain insight into the maternal and zygotic transcriptomes. De novo assembly of reads obtained from the egg and late larval stages generated 86,898 transcripts and recovered more than 95 % of the predicted genes stored in the OikoBase. These results confirmed and expanded the results of tiled microarray analysis by Danks et al. (Danks et al. 2013), which detected 77 % of the predicted genes (13,081 out of 16,749 tested genes) at several time points from egg to adult. The depth of the sequencing data showed three novel aspects: (1) 175 novel protein-encoding genes were identified; 31 of which were not found in the Norwegian genome reference. (2) Transcriptome-wide comparison revealed high levels of sequence variation (∼10 % of nucleotides in the intragenic region were different) between the two O. dioica populations. (3) Trans-splicing tended to happened less in larva-specific mRNAs when compared with egg mRNAs.
O. dioica is characterized by rapid development, organogenesis being completed within 10 h post-fertilization (hpf) to form a functional body that is a miniature of the adult (Nishida 2008). In this study, we collected samples at only two stages: the eggs and the 8 hpf larvae. Sampling of these two developmental stages was enough to recover almost all of the predicted genes (16,423 out of 17,212 OikoBase genes) and EST clones (101,270 out of 103,969). This contrasts with the case of vertebrates, for which about 86 % of genes were recovered even when samples were collected from 6 to 10 different stages (Aanes et al. 2011).
In addition, we identified 175 novel protein-encoding genes. The number of O. dioica genes is estimated to be approximately 18,000 in the genome reference assembly (Denoeud et al. 2010; Seo et al. 2001). Therefore, about 1 % of genes have not been annotated in the genome reference. The 175 novel protein-encoding genes that possess Pfam domains did not show any hit with predicted proteins in OikoBase and other public protein databases (UniProtKB/Swiss-Prot and NCBI RefSeq databases) by BLASTX, but their expression was confirmed by RT-PCR. Intriguingly, 31 of the novel genes were not found in the genome reference derived from the Norwegian O. dioica. There are two possible explanations for this, the first being lower coverage of the genome reference. The genome sequence of O. dioica was determined by traditional shotgun sequencing, and the average genome coverage was 14X, and 3 % of ESTs were not mapped onto the genome reference (Denoeud et al. 2010; Seo et al. 2001). Therefore, some genes might be missing in the genome reference. The other possible reason is divergence of the genome sequences between the Japanese and Norwegian populations.
O. dioica is considered to be a rapidly evolving chordate because of its short life cycle of 5 days and rapid changes in its genome organization (Delsuc et al. 2006). However, no previous studies have tested intra-species sequence divergence in appendicularian species. In the present study, the transcript sequences showed a high degree of variability between the Japanese and Norwegian O. dioica populations. The average degrees of nucleotide and amino acid sequence conservation were 91.0 and 94.8 %, respectively. The actual SNP rate would be higher, since we did not count gaps and eliminated the highly unmatched sequences in the comparison. It is noteworthy that the percentage differences in the sequences were comparable to those between two cryptic species of C. intestinalis, i.e., type A and type B, in which reproductive isolation has become established (Caputi et al. 2007; Nydam and Harrison 2010; Suzuki et al. 2005); the sequence similarities in the protein-encoding regions were 87–98 % at the nucleotide level and 93.4–100 % at the amino acid level, depending on the genes compared (Caputi et al. 2007; Nydam and Harrison 2010; Suzuki et al. 2005). These facts suggest that whole exons in the genome, i.e., the exome, have become highly diverged between geographically distant O. dioica populations, although it is not known whether hybrids of the Japanese and Norwegian O. dioica would be fertile or infertile. Future analysis of the genome of the Japanese O. dioica would be warranted to gain insight into the genomic plasticity of this rapidly evolving metazoan.
Among the 12,311 assembled transcripts, 63 and 99 % were detected in eggs and larvae, respectively. Thus, the mRNAs of most of genes are present at the developing larval stage. In this quick developer, it is most probable that residual maternal mRNAs are still preserved in 8 hpf larvae. Quantitative analysis indicated that 5108 genes were up-regulated (fold change ≥4) or down-regulated (fold change ≤−4) between the two stages, accounting for 41 % of the total unigenes. GO enrichment analysis showed that the up-regulated and down-regulated genes were linked to distinct biological processes.
In O. dioica, 145 trans-spliced mRNAs have been found from 1155 EST clones, and at least 25 % of the mRNAs were estimated to be trans-spliced (Ganot et al. 2004). Detection of SL sequences among whole EST sequences has revealed that ∼30 % of the genes are trans-spliced (Denoeud et al. 2010). In the present analysis, the SL was observed in 40.8 % of mRNA species. It showed preferential linkage to adenine at the 5′ ends of the downstream exons. Intriguingly, the trans-splicing occurs more frequently in eggs than in larvae. SL was found in 20.6 % of the down-regulated genes, whereas it was found in only 8.4 % of the up-regulated genes. Gene with SL showed similar GO enrichment with the case of the down-regulated genes. These results confirm and well coincide with the recent paper (Danks et al. 2015), which has shown that maternal transcripts tend to be more frequently trans-spliced in O. dioica. These results raise an interesting possibility that trans-splicing occurs more frequently in maternal mRNAs than zygotic ones, and it plays roles in early embryogenesis.
Conclusions
In conclusion, our data have created a transcriptome resource for the Japanese O. dioica. Our results support the in silico prediction of gene products in the genome of the Norwegian O. dioica (Danks et al. 2013). Furthermore, the depth of RNA-Seq analysis clarified various new aspects of the O. dioica transcriptome. First, 175 novel protein-encoding genes were found. Second, transcriptome-wide comparison revealed high levels of exon sequence variation between the Japanese and Norwegian populations. Finally, analysis of SL suggested that trans-splicing occurs more frequently in eggs than in larvae. The present results will provide an additional resource that is useful for understanding the developmental processes and evolutional aspects of this chordate.
FC, Fold change; GO, Gene ontology; Hpf, Hours post-fertilization; SL, Spliced leader
References
Aanes H et al (2011) Zebrafish mRNA sequencing deciphers novelties in transcriptome dynamics during maternal to zygotic transition. Genome Res 21:1328–1338. doi:10.1101/gr.116012.110
Adiconis X et al (2013) Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat Methods 10:623–629. doi:10.1038/nmeth.2483
Bassham S, Postlethwait J (2000) Brachyury (T) expression in embryos of a larvacean urochordate, Oikopleura dioica, and the ancestral role of T. Dev Biol 220:322–332. doi:10.1006/dbio.2000.9647
Bauer S, Grossmann S, Vingron M, Robinson PN (2008) Ontologizer 2.0–a multifunctional tool for GO term enrichment analysis and data exploration. Bioinformatics 24:1650–1651. doi:10.1093/bioinformatics/btn250
Bouquet JM, Spriet E, Troedsson C, Ottera H, Chourrout D, Thompson EM (2009) Culture optimization for the emergent zooplanktonic model organism Oikopleura dioica. J Plankton Res 31:359–370. doi:10.1093/plankt/fbn132
Boutet E, Lieberherr D, Tognolli M, Schneider M, Bairoch A (2007) UniProtKB/Swiss-Prot. Methods Mol Biol 406:89–112
Burge SW et al (2013) Rfam 11.0: 10 years of RNA families. Nucleic Acids Res 41:D226–D232. doi:10.1093/nar/gks1005
Caputi L, Andreakis N, Mastrototaro F, Cirino P, Vassillo M, Sordino P (2007) Cryptic speciation in a model invertebrate chordate. Proc Natl Acad Sci U S A 104:9364–9369. doi:10.1073/pnas.0610158104
Danks G et al (2013) OikoBase: a genomics and developmental transcriptomics resource for the urochordate Oikopleura dioica. Nucleic Acids Res 41:D845–D853. doi:10.1093/nar/gks1159
Danks GB, Raasholm M, Campsteijn C, Long AM, Manak JR, Lenhard B, Thompson EM (2015) Trans-splicing and operons in metazoans: translational control in maternally regulated development and recovery from growth arrest. Mol Biol Evol 32:585–599. doi:10.1093/molbev/msu336
Delsuc F, Brinkmann H, Chourrout D, Philippe H (2006) Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature 439:965–968. doi:10.1038/nature04336
Denoeud F et al (2010) Plasticity of animal genome architecture unmasked by rapid evolution of a pelagic tunicate. Science 330:1381–1385. doi:10.1126/science.1194167
Eddy SR (2011) Accelerated profile HMM searches. PLoS Comput Biol 7:e1002195. doi:10.1371/journal.pcbi.1002195
Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinf 5:113. doi:10.1186/1471-2105-5-113
Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150–3152. doi:10.1093/bioinformatics/bts565
Fujii S, Nishio T, Nishida H (2008) Cleavage pattern, gastrulation, and neurulation in the appendicularian, Oikopleura dioica. Dev Genes Evol 218:69–79. doi:10.1007/s00427-008-0205-4
Ganot P, Kallesoe T, Reinhardt R, Chourrout D, Thompson EM (2004) Spliced-leader RNA trans splicing in a chordate, Oikopleura dioica, with a compact genome. Mol Cell Biol 24:7795–7805. doi:10.1128/MCB.24.17.7795-7805.2004
Gasparini F, Shimeld SM (2011) Analysis of a botryllid enriched-full-length cDNA library: insight into the evolution of spliced leader trans-splicing in tunicates. Dev Genes Evol 220:329–336. doi:10.1007/s00427-011-0351-y
Grabherr MG et al (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652. doi:10.1038/nbt.1883
Griggio F et al (2014) Ascidian mitogenomics: comparison of evolutionary rates in closely related taxa provides evidence of ongoing speciation events. Genome Biol Evol 6:591–605. doi:10.1093/gbe/evu041
Haas BJ et al (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8:1494–1512. doi:10.1038/nprot.2013.084
Harris MA et al (2004) The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res 32:D258–D261. doi:10.1093/nar/gkh036
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36. doi:10.1186/gb-2013-14-4-r36
Kishi K, Onuma TA, Nishida H (2014) Long-distance cell migration during larval development in the appendicularian, Oikopleura dioica. Dev Biol 395:299–306. doi:10.1016/j.ydbio.2014.09.006
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf 12:323. doi:10.1186/1471-2105-12-323
Li W, Godzik A (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659. doi:10.1093/bioinformatics/btl158
Matsumoto J et al (2010) High-throughput sequence analysis of Ciona intestinalis SL trans-spliced mRNAs: alternative expression modes and gene function correlates. Genome Res 20:636–645. doi:10.1101/gr.100271.109
Nawrocki EP, Eddy SR (2013) Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29:2933–2935. doi:10.1093/bioinformatics/btt509
Nishida H (2008) Development of the appendicularian Oikopleura dioica: culture, genome, and cell lineages. Dev Growth Differ 50(Suppl 1):S239–S256. doi:10.1111/j.1440-169X.2008.01035.x
Nishida H, Stach T (2014) Cell lineages and fate maps in tunicates: conservation and modification. Zoolog Sci 31:645–652. doi:10.2108/zs140117
Nydam ML, Harrison RG (2010) Polymorphism and divergence within the ascidian genus Ciona. Mol Phylogenet Evol 56:718–726. doi:10.1016/j.ympev.2010.03.042
Omotezako T, Nishino A, Onuma TA, Nishida H (2013) RNA interference in the appendicularian Oikopleura dioica reveals the function of the Brachyury gene. Dev Genes Evol 223:261–267. doi:10.1007/s00427-013-0438-8
Pruitt KD, Tatusova T, Maglott DR (2005) NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 33:D501–D504. doi:10.1093/nar/gki025
Rice P, Longden I, Bleasby A (2000) EMBOSS: the European molecular biology open software suite. Trends Genet 16:276–277. doi:10.1016/S0168-9525(00)02024-2
Satou Y, Hamaguchi M, Takeuchi K, Hastings KE, Satoh N (2006) Genomic overview of mRNA 5′-leader trans-splicing in the ascidian Ciona intestinalis. Nucleic Acids Res 34:3378–3388. doi:10.1093/nar/gkl418
Seo HC et al (2001) Miniature genome in the marine chordate Oikopleura dioica. Science 294:2506. doi:10.1126/science.294.5551.2506
Shirae-Kurabayashi M, Nishikata T, Takamura K, Tanaka KJ, Nakamoto C, Nakamura A (2006) Dynamic redistribution of vasa homolog and exclusion of somatic cell determinants during germ cell specification in Ciona intestinalis. Development 133:2683–2693. doi:10.1242/dev.02446
Stach T, Winter J, Bouquet JM, Chourrout D, Schnabel R (2008) Embryology of a planktonic tunicate reveals traces of sessility. Proc Natl Acad Sci U S A 105:7229–7234. doi:10.1073/pnas.0710196105
Suzuki MM, Nishikawa T, Bird A (2005) Genomic approaches reveal unexpected genetic divergence within Ciona intestinalis. J Mol Evol 61:627–635. doi:10.1007/s00239-005-0009-3
Vandenberghe AE, Meedel TH, Hastings KE (2001) mRNA 5′-leader trans-splicing in the chordates. Genes Dev 15:294–303. doi:10.1101/gad.865401
Wu B et al (2012) Comprehensive transcriptome analysis reveals novel genes involved in cardiac glycoside biosynthesis and mlncRNAs associated with secondary metabolism and stress response in Digitalis purpurea. BMC Genomics 13:15. doi:10.1186/1471-2164-13-15
Xie Y et al (2014) SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 30:1660–1666. doi:10.1093/bioinformatics/btu077
Acknowledgments
We appreciate M. Suzuki, M. Hayashi, and M. Isobe in our laboratory for their help in the culture of O. dioica. We thank the Genome Information Research Center in Osaka University for providing computational resources.
Funding
This work was supported by Grants-in-Aid for Scientific Research from the JSPS to H.N. (22370078, 26650079) and T.A.O. (22870019, 26840079). K.W. is supported by a MEXT Scholarship (125058), a Mitsubishi Corporation International Scholarship (MITSU1451), and an Osaka University Scholarship and Research Assistant Fellowship.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KW, HN, and TAO designed the study and wrote and revised the manuscript. KW performed all bioinformatics analysis. TO, KK, and TAO carried out sample collection and experiments. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by Volker G. Hartenstein
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table S1
Primers used for RT-PCR analyses. (DOCX 21 kb)
Supplementary Table S2
Comparison of SOAPdenovo-Trans and Trinity de novo assembly. (DOCX 19 kb)
Supplementary Table S3
List of the total 12,311 known and novel unique protein-encoding unigenes that were sorted by the gene ID with their level of expression in eggs and 8 hpf larvae (FPKM). (XLSX 939 kb)
Supplementary Table S4
Expression (FPKM) of three genes with already well-known patterns. Brachyury and Muscle actin are typical zygotic genes, while Vasa is a maternally expressed gene. (DOC 30 kb)
Supplementary Table S5
Gene ontology annotation of the 3772 up-regulated and 1336 down-regulated genes. Terms with Bonferroni adjusted p value of ≤0.01 were selected. (XLSX 20 kb)
Supplementary Table S6
Comparison of the function of unigenes with SL and that without SL. Terms with Bonferroni adjusted p value of ≤0.01 were selected. (XLSX 19 kb)
Rights and permissions
About this article

Cite this article
Wang, K., Omotezako, T., Kishi, K. et al. Maternal and zygotic transcriptomes in the appendicularian, Oikopleura dioica: novel protein-encoding genes, intra-species sequence variations, and trans-spliced RNA leader. Dev Genes Evol 225, 149–159 (2015). https://doi.org/10.1007/s00427-015-0502-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00427-015-0502-7