
Abstract
Mutations in the lozenge gene of Drosophila melanogaster elicit a pleiotropic set of adult phenotypes, including severe compound eye perturbations resulting from the defective recruitment of photoreceptors R1/6 and R7, cone and pigment cells. In this study, we show that excessive patterned apoptosis is evident at the same developmental stage in these lozenge mutants. In lozenge null mutants, apoptosis occurs prior to lozenge-dependent cell fate specification. A second gene, D-Pax2, genetically interacts with lozenge. Interestingly, D-Pax2 mutants also exhibit increased cell death, but slightly later in development than that in lozenge mutants. Although expression of the caspase inhibitor p35 eliminates death in both lozenge and D-Pax2 mutants, the lozenge mutant eye phenotypes persist because other normal Lozenge functions are still lacking. D-Pax2 eye phenotypes, in contrast, are dramatically altered in a p35 background, because cells that normally differentiate as cone and primary pigment cells are subsequently transformed into secondary pigment cells. This study leads us to propose that Lozenge, aside from its known role in gene regulation of cell-specific transcription factors, is required to contribute to the repression of cell death mechanisms, creating a permissive environment for the survival of undifferentiated cells in early eye development. Lack of lozenge expression increases the likelihood that an undifferentiated cell will initiate its default death program and die prematurely. The ectopic cell death evident in D-Pax2 mutants appears to arise from the cell fate transformation of cone cells into secondary pigment cells, either autonomously or as a result of defective signalling.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Programmed cell death, or apoptosis, plays an important role in the patterning of emerging adult structures. In development, cells are first organised into a loose framework surrounded by excess cells. Apoptosis then removes excess cells in order to sculpt emerging structures (reviewed in Rusconi et al. 2000). This process must be co-ordinated with other developmental events, including cell division, migration and differentiation (reviewed in Bonini and Fortini 1999). Apoptosis has even been proposed to be a "default program" in development, in order to minimise the risk of cells proliferating uncontrollably (McNeill and Downard 1999). If this is the case, then cells must employ survival signals to control apoptosis.
Apoptosis occurs through activation of highly conserved molecular pathways. Elements of cell death machinery, such as CED-3-related cysteine proteases (caspases), have been conserved in nematodes, flies and mammals (Meier and Evan 1998). Furthermore, some caspase inhibitors including the baculovirus protein p35 can prevent apoptosis in all these species (Hay et al. 1994; Rabizadeh et al. 1993). In Drosophila, three genes, reaper (rpr), grim and head involution defective (hid), are required to activate apoptosis (reviewed in Abrams 1999; Bangs and White 2000). Without these genes, normal apoptosis in embryonic development cannot occur and embryos die (White et al. 1994). Conversely, ectopic expression of any of these genes results in excess apoptosis through activation of a caspase pathway (Chen et al. 1996; White et al. 1996). rpr and Grim are transcribed only in cells doomed to die, while hid is expressed in both cells that survive and in those that die (White et al. 1994; Grether et al. 1995). The mechanisms of regulation for grim and rpr remain mostly unknown, while hid appears to be down-regulated by the Ras/MAP kinase pathway, both at the level of Hid phosphorylation and of hid expression (Bergmann et al. 1998; Kurada and White 1998). Recently it has been shown that EGFR signalling prevents apoptosis by inactivation of hid expression or function in the Drosophila eye (Yu et al. 2002).
Apoptosis is a key component of Drosophila visual system development (Wolff and Ready 1991). The compound eye is comprised of approximately 800 ommatidia. Each ommatidium contains eight photoreceptor neurons and a collection of supporting cells. The eye begins developing in the eye imaginal disc epithelium at mid-third larval instar, with the morphogenetic furrow progressing anteriorly across the disc, marking the onset of cell differentiation and pattern formation (Ready et al. 1976). Photoreceptor R8 is the first cell established, its recruitment mediated by signalling events co-ordinated by the furrow. R2/5/3 and R4 are then added to create the five-cell pre-cluster (Wolff et al. 1997). Unspecified cells in this region then undergo synchronous mitosis to re-populate the epithelium (Ready et al. 1976; Finley et al. 1996). The remaining photoreceptors (R1/6/7), cone and pigment cells are then added. Normally, minimal cell death occurs during third larval instar eye development. However, apoptosis is a major developmental event in the pupal retina, removing cells that have failed to establish a fate after fates of all requisite cells have been defined (Wolff and Ready 1991).
Many genes play roles in fate specification and ommatidial assembly in Drosophila eye development, including both lozenge (lz) and D-Pax2. Lz, a nuclear protein, is a member of the Runx (runt/Aml1) family of transcription factors (Daga et al. 1996). The use of a lz gal4;UAS-GFP enhancer trap line and antibody staining has revealed the expression of lz in undifferentiated non pre-cluster cells immediately posterior to the furrow, in R1/6/7 neurons, and in cone and pigment cells, but not in developing bristle cells (Flores et al. 1998; Crew et al. 1997). Mutations in lz result in defects in the number and identity of photoreceptor cells per ommatidium, and also in cone and pigment cell structures (Crew et al. 1997). Lz is a contributor to cell fate establishment in the eye, partaking in the regulation of genes including Bar, seven-up, D-Pax2, prospero, and dpn (Daga et al. 1996; Crew et al. 1997; Canon and Banerjee 2000; Flores et al. 2000; Xu et al. 2000). D-Pax2 [originally named sparkling (spa)] is also involved in fate specification in the eye (Lindsley and Zimm 1992; Fu et al. 1998). D-Pax2 mutants exhibit pigment and cone cell defects (Lindsley and Zimm 1992). Beeson and Bender (1975) originally showed that spae (lz) enhances lz mutant eye phenotypes, suggesting that lz and D-Pax2 gene products interact. Recently it has been shown that Lz, along with other factors, contributes to the regulation of D-Pax2 in a context-dependent manner (Flores et al. 2000).
Ectopic cell death has been observed in the developing eye of Drosophila mutants exhibiting eye defects, including Bar, rough, eyes absent and yan. Cell mis-specification, or failure of cell differentiation can lead to death of aberrant cells during the third instar, giving rise to these eye phenotypes. It is equally possible that if apoptosis is a "default program" in development, and cells employ survival signals to shut off the initiation of the death cascade, then removal of these signals will result in ectopic apoptosis. Survival signals have recently been identified in Drosophila eye development in EGFR signalling through the receptor and through its ligand Spitz and possibly Ras/MAP kinase signalling (Bergmann et al. 1998; Dominguez et al. 1998; Kurada and White 1998; Baker and Yu 2001; Yu et al. 2002).
In this study, we investigated the effect of lz and D-Pax2 mutations on apoptosis in early eye development. Patterned apoptosis was observed posterior to the furrow in all mutants. However, in lz null mutants, death is evident before lz-dependent R1/6/7 neuronal and cone cells have been added to the developing ommatidial clusters. This suggests that neither failure of cell differentiation nor cell mis-specification is the cause of ectopic death. Mutations in D-Pax2 cause ectopic death to occur later than in lz mutants and via a different mechanism.
We propose that the initial expression of lz in the developing eye provides a survival signal by repressing cell death. Secondary expression of lz after the second mitotic wave is required for correct cell fate specification. The cell death evident in D-Pax2 mutants appears to arise from the cell fate transformation of cone cells into secondary pigment cells, either autonomously or as a result of defective signalling.
Materials and methods
Fly strains and genetics
Most lz stocks were obtained from the Bloomington Stock Center, Indiana, Mel Green (University of California at Davis) and Reinhard Stocker (University of Fribourg, Switzerland). lzr1 and lzr7 were provided by Dr. U. Banerjee. pGMR-p35 flies were provided by Gerald Rubin (University of California at Berkeley). Wild-type, attached-X and balancer chromosome stocks were obtained from the Bowling Green Stock Center, Ohio, or from Bloomington and are described in Lindsley and Zimm (1992). Flies were reared on standard corn meal molasses or standard laboratory medium at 25°C. lz ts1 was also reared at 29°C. White pre-pupae were selected by morphology and designated as 0- to 3-h-old pre-pupae by selecting those that sank in 1 M NaCl (Pollock et al. 1990).
Histology, immunohistochemistry and microscopy
Toluidine blue staining was achieved as described in Batterham et al. (1996). For transmission electron microscopy, white pre-pupal eye-antennal discs were prepared as described in Baumann and Walz (1989). Sections of 0.5 µm were cut, stained and viewed on a JEOL 4000EX (400 keV) electron microscope at the National Center for Microscopy and Imaging Research at San Diego (M.H. Ellisman, Director).
Scanning electron microscopy of 0- to 3-day-old flies was achieved as described in Batterham et al. (1996). Images were digitally acquired using Spectrum software. Alternatively, unfixed 0- to 3-day-old flies were imaged using a Hitachi S-2460N.
Acridine orange (AO) staining was achieved as described in Spreij (1971). In some cases, tissue was double stained with Hoechst dye (33258).
TUNEL labelling was achieved using Roche's In situ Cell Death Detection Kit (fluorescein), with the protocol modified as follows: third instar eye discs were first fixed in 4% paraformaldehyde in PBS, washed and permeabilised in 0.2% Triton-X in PBS, then incubated in TUNEL mixture at 37°C as per manufacturers protocol. Discs were stained further as appropriate. Rat anti-Elav (O'Neill et al. 1994) was detected with anti-Rat Alexa594 (Molecular Probes). The Optiscan F900e confocal system with an Olympus BX60 microscope was used to image and examine samples. Images were processed using Adobe Photoshop 6.0 software.
In situ hybridisation
A plasmid containing a rpr cDNA (clone 13B2) was provided by Dr. H. Steller. The rpr cDNA was random prime labelled with digoxigenin-dUTP (Boehringer Mannheim). Eye discs and control embryos were probed as described in Tautz and Pfeiffle (1989).
Molecular biology
Total RNA was isolated using the RNeasy mini kit (Qiagen) following manufacturers instructions. Forty to fifty Canton S and lz mr1 white pre-pupal cephalic complexes (brain lobes, eye-antennal disks, and mouth hooks) were dissected in PBS and held on ice.
cDNA was synthesised from 8 µl RNA using the 3′ RACE system for rapid amplification of cDNA ends (Invitrogen) following manufacturer's instructions.
Three replicates of quantitative fluorescence detection PCR were achieved using methods similar to those in Reja et al. (2002). Conditions were 1 cycle of 10 min at 95°C and 55 cycles at 95°C for 20 s, 63°C for 20 s, and 72°C for 85 s. SYBR green fluorescence was measured after each extension step in a Corbett Research Rotor-Gene Real-Time Thermal Cycler. Forward: Saba2 5′CCGTCGATGGACCCCGCGAG3′ and reverse: Saba1 5′TTGGGTCAATCGGGTCGCCACAC3′ primers amplified a 1,294-bp product from exons 4 to 6. Canton S and lz mr1 cDNA was tested with exon 5 primers producing a 387-bp product (forward: 5F 5′CCACGACTCCGCCACAGGTG3′ and reverse: 5R 5′CGAGTTGATTGTACTATTCG3′) and with exon 6 primers giving a 748-bp product (forward: 6F 5′CGGCGAATGGACAAATGGATCCTCG3′ and reverse: Saba1). Real-time PCR of β-actin (Fisher-Biotech, Australia) used defined cDNA and primers to produce a 180-bp amplicon. Conditions were 1 cycle for 120 s at 95°C and 45 cycles at 95°C for 20 s, 63°C for 20 s and 72°C for 30 s. The β-actin amplification took off at cycle 30 (6,000 copies), cycle 33 (600), and cycle 36 (60) showing that cycle doubling was true.
Standard PCR and sequencing reactions consisted of hot start PCR Promega reagents following manufacturers protocol in a Mastercycler gradient (Eppendorf) or Progene (Techne) thermocycler. Sequencing chemistry used ABI Prism Big Dye Terminator (version 2) Cycle Sequencing Ready Reaction Kit (Perkin-Elmer). Sequence reactions were run and detected on gels by the Australian Genome Research Facility and results analysed with Sequencher 3.1 (Gene Codes Corporation).
lz r1 , lz r7 and lz L deletions were mapped via PCR walking at the ends of potential breakpoints and across breakpoints to identify 5′ and 3′ limits. lz mr1 has been previously characterised (Daga et al. 1996; Flores et al. 1998; Behan et al. 2002). Insertion mutations were localised by long PCR using the Expand 20KbPlus PCR system (Boehringer Mannheim) with genomic DNA-specific primers. Long PCR products were purified and sequenced to determine exact breakpoints and sequence identity of the element.
Results
Mutations in lozenge invoke ectopic cell death in early eye development
lz mutant eye phenotypes vary in severity from being mild to severe (Fig. 1b–e). Retinal sections of severe mutants highlight pigment cell, fenestrated membrane, and ommatidial structure abnormalities (Fig. 1g–j), along with defects in the identity and number of photoreceptors per cluster (Fig. 1g). These observations correlate with previous data showing that neurons R1/6 and R7, and non-neuronal cone and pigment cells are all affected in lz mutants (Crew et al. 1997). To determine whether ectopic cell death is a phenotype attributable to lz mutants, strains were analysed for the presence of excessive acridine orange (AO) staining in early eye development (Fig. 1, Table 1). Analysis highlighted excess AO staining in mutants compared to the wild-type strain (Fig. 1k–o). Furthermore, a distinct, synchronous pattern of staining posterior to the furrow was observed (Fig. 1l, m, o).

Adult and developing eye phenotypes of wild-type and lozenge (lz) mutants. Scanning electron micrographs (SEMs) of wild-type (a) and lz mutants (b–e). The temperature-sensitive mutant, lz ts1, was reared at both permissive (d; 18°C) and restrictive temperatures (e; 29°C). f–j. Toluidine-blue-stained 1-μm retinal sections (arrows show photoreceptor rhabdomeres; L lens, br brain). In lz mr1(g), ommatidia have too few cells (white arrow), or extra cells (inset). In lz L (h), clusters are rarely identifiable (black arrows). Sections of lz ts1 at 18°C are normal (i); lens and fenestrated membrane defects are observed at 29°C (j). k–o. Live eye imaginal discs stained with acridine orange (AO; arrow shows furrow). In lz ts1 mutants, staining increases proportionally to eye severity (n–o). Other lz discs (l–m) exhibit excessive, patterned cell death compared to minimal death in wild-type (k)
To determine whether ectopic death was only a feature of severe mutants, 26 lz strains, varying in eye severity, were assayed for AO staining. Results showed excess staining in mild and severe mutants, with intensity increasing proportionally to phenotypic severity (Table 1). Ectopic death could not be attributed to the genetic background, since the lz mr1 mutant continued to exhibit excess cell death after it was outcrossed for 12 generations to a wild-type strain that showed minimal cell death (Fig. 1k).
Further evidence supporting the link between ectopic cell death and lz mutations comes from analysis of the temperature-sensitive mutant, lz ts1. The lz ts1 eye appears normal at permissive temperatures and exhibits minimal death in discs (Fig. 1d, i, n). At restrictive temperatures, the eye is moderately perturbed and excessive cell death is observed (Fig. 1e, j, o). Additional experiments also showed that when lz eye phenotypes are enhanced or reverted, a subsequent increase or decrease in cell death is observed (Table 1).
These results reveal that ectopic cell death observed in lz eye discs is a direct result of mutations in the lz gene. Other experiments have further revealed that elevated cell death is specific to imaginal tissues affected by lz mutations (not shown).
Ectopic cell death occurs in a patterned wave early in eye development of lz mutants, prior to photoreceptor differentiation
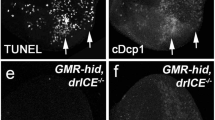
Initial developmental events associated with furrow progression and recruitment of the five-cell pre-cluster (R8/2/5/3/4) proceed normally in lz mutants (Crew et al. 1997). To establish the timing of ectopic death relative to these events, TUNEL (terminal deoxynucleotide transferase TdT-mediated dUTP nick labelling) analysis was coupled with cell-specific antibody staining in lz third instar eye discs. TUNEL preferentially labels apoptotic cells by incorporating labelled nucleotides to DNA strand breaks (Gold et al. 1994). Excessive apoptosis was observed in a synchronised pattern posterior to the furrow (Fig. 2b, c). Interestingly, the onset of death is earlier in the lz null (lz L) compared to the severe mutant (lz mr1). In lz L, cell death is observed between rows 3 and 7 posterior to the furrow (Fig. 2c), while in lz mr1, apoptosis is observed between rows 7 and 12 (Fig. 2b). This difference is highlighted further when TUNEL is coupled with a mitosis marker. In lz mr1, death occurs after the synchronous round of mitosis with no observable overlap (Fig. 2e). Conversely, the ectopic death in lz L slightly overlaps this mitotic event (Fig. 2f).

Immunohistochemistry and TEMs show the pattern and timing of apoptosis in wild-type and lozenge mutants. a–c TUNEL (green) of apoptotic cells and anti-ELAV staining (red) of neuronal cells in wild-type and lz third instar eye discs (arrow marks furrow; posterior towards bottom in this and subsequent figures). Wild-type (a) shows minimal apoptosis. All eight photoreceptors (red) are observed in specific apical positions observable in specific planes using confocal microscopy (insets). lz mutants (b, c) show increased apoptosis (green) beginning later (posterior to the furrow) in lz mr1 (b) than in lz L (c). Only five photoreceptors (R8/2/5/3/4) are observed in the mutants (red; inset in b only). d–f. TUNEL (green) and anti-phospho-histone H3 mitosis marker (red) in wild-type and lz discs. Mitosis is normal in lz discs (e, f), comparative to wild-type (d). Mitotic cells are observed anterior and posterior (second mitotic wave) to the furrow. Death is more posterior in lz mr1 (e) than in lz L (f). g–i. Transmission electron micrographs confirm that ectopic apoptosis occurs in lz mutants. For g–i, apical microvilli are at the top, posterior to the right (see j for orientation of frame). Condensed, osmophilic nuclei indicate apoptosis. High magnification of the base of wild-type discs shows occasional dying cells in lower half of disc (g). In lz mr1 and lz L (h, i), cell death is evident from the apical to basal surface. j Low magnification view of the wild-type eye discs reveals the frame used in g. The black arrowhead indicates the position of the furrow just below the peripodial membrane. The basal side (bottom) has neurites growing toward the optic stalk (bottom right corner)
The specification of photoreceptors R1/6/7 in eye development is dependent on lz expression (Daga et al. 1996; Crew et al. 1997; Flores et al. 1998; Behan et al. 2002). These cells are recruited to developing ommatidia after the second mitotic round, approximately four to five rows posterior to the furrow (Tomlinson and Ready 1987). One explanation for the ectopic death observed in lz mutants is that these cells die because their fates could not be established. However, in lz null mutants, the pattern of TUNEL labelling indicates that apoptosis is occurring before R1/6/7 and cone cells are added to developing ommatidia (Fig. 2). To further gauge whether cells have established a fate before they die, transmission electron micrographs of eye disc cross-sections were analysed for the presence of apoptotic cells (Fig. 2g–i). Several hallmarks of apoptosis, including condensation of the cell nucleus with otherwise intact organelles, fragmentation of dying cells, and an increased osmophilicity (Wolff and Ready 1991) were present in lz mutants (Fig. 2h–i). Interestingly, wild-type discs showed that a small amount of apoptosis occurs to eliminate cells that have not divided correctly. These rare, dying cells reside near the basal portion of the disc (Fig. 2g, j). In lz mutants, the fragmented dead cells are found in apical positions (Fig. 2h, i). Nuclear position is a key indicator of the developmental state of cells in the eye disc, as differentiating cells make a dramatic shift to position the nucleus near the apical margin of the epithelium (Tomlinson and Ready 1987). The apical position of dying cells in lz mutant discs indicates that apoptotic cells were in the process of establishing a fate and undergoing concomitant nuclear movements prior to their deaths. Taken together, both TUNEL analysis and TEM studies reveal that cells are dying prior to lz-dependent R1/6 and R7 cell fate specification.
Onset of apoptosis in lz mutants is allele specific
Differing patterns of death were revealed in a lz null and in a loss-of-function mutant. Consequently, other lz alleles were surveyed for their cell death pattern. Long PCR and sequencing of lz has elucidated both the position and nature of mutations (Fig. 3a). Corresponding TUNEL analysis (Fig. 3b–i) showed that both null mutants (Fig. 3d, h lz L , e, i lz r1) exhibit cell death at an earlier developmental stage relative to lz mr1(Fig. 3b, f) and lz r7 (Fig. 3c, g). These latter two mutants result from smaller deletions (Fig. 3a) and exhibit a pattern of TUNEL labelling reflective of a later onset of death.

a Molecular map of the lozenge (lz) gene indicating the lesions for the mutant alleles. Shaded boxes represent the six exons of the 3.5-kb transcriptional isoform of the lz gene (c3.5). Each of the mutations analysed are deletions. The precise mutational breakpoints can be positioned according to GenBank entry AF217651. The lz L (null) deletion spans from within intron 2 to exon 6, precisely 5′ after 9,798 bp and 3′ before 26,295 bp. This mutant also has a LTR-like retrotransposon (approximately 8 kb) inserted into this position (*). lz r1 (null) is a deletion spanning most of the gene, beginning 5′ after position 8,290 and ending 3′ before position 31,007. lz mr1 deletes the eye-specific enhancer (intron 2) between positions 9,585 (5′) and 10,983 (3′). The lz r7 deletion spans from exons 4 to 6, 5′ after 15,353 bp and 3′ before 29,135 bp. b–e TUNEL with anti-ELAV staining in lz mutants (arrow shows furrow). Apoptosis (green) in nulls (lz L and lz r1; d, e) is earlier than in lz mr1 and lz r7 (b, c). This observation is reaffirmed in TUNEL and anti-phospho-histone H3 experiments (f–i). j Real time RT-PCR of mRNA from lz mr1 and wild-type white pre-pupal discs (cycle number on x axis, fluorescence detected on y). Three replicates of each sample were amplified for 50 cycles with primer pairs Saba1 and Saba2 encompassing exon 4 to exon 6. A two to three cycle separation between the amplification phase for wild-type (red) versus lz mr1 (blue) indicates that the lz mr1 transcript is produced at less than 10% of the normal level. Comparable results were achieved with primers within exon 5 (5F and 5R) and within exon 6 (6F and Saba1; see Materials and methods)
Since lz null mutants (lz L and lz r1) produce no lz transcript due the nature of their mutations (Fig. 3a), quantitative fluorescence detection reverse transcriptase PCR from white pre-pupal eye discs was used to assess whether a transcript is produced in lz mr1. lz mr1 was examined because it has a deletion in the eye-specific enhancer (intron 2), but all exons remain intact (Fig. 3a). Results showed that both wild type and lz mr1 produce transcripts, but the lz mr1 transcript is produced at less than 10% of that of the wild type (Fig. 3j). This conclusion can be made based on the specific onset of amplification of the gene product in the lz mr1 mutant being later than that in the wild type. Sequence analysis of a PCR product from lz mr1 confirmed that it was a lz transcript (not shown). Control standards of a serial dilution of β-actin cDNA confirmed the doubling of the PCR reaction in real-time analysis (not shown).
These results show that removing all the lz gene product has a more severe effect on the mechanism and timing of death than reducing normal levels of lz. Such a null mutation results in apoptotic death occurring before lz dependent R1/6/7 and cone cells have been added to the developing ommatidial clusters in third instar discs, indicating the necessity of lz gene product for early cell survival.
reaper expression is elevated in lz mutant eye discs
Expression of rpr has been described as a prelude to cell death, rpr being both necessary and sufficient to induce cell death in Drosophila embryos (White et al. 1994; McCall and Steller 1997). Analysis of rpr expression was carried out in lz mutants using whole-mount in situ hybridisation to eye discs (Fig. 4). Very little rpr expression is evident in the wild-type eye (Fig. 4a). In contrast, lz mr1 discs (Fig. 4b) exhibit excessive rpr-positive cells, rpr expression beginning very close to the furrow and several rows ahead of the cell death observed with TUNEL labelling. This is consistent with the known delay in rpr action (White et al. 1994; McCall and Steller 1997).

In situ hybridisation with a digoxigenin-labelled reaper (rpr) cDNA on wild-type and lz mr1 discs. Wild-type exhibits little rpr expression (a; arrowhead marks furrow, arrow shows rpr expression enlarged in inset). lz mr1 (b) exhibits increased rpr expression close to the furrow. c–j. SEMs and immunohistochemistry show that p35 expression blocks apoptosis in lz mutants, but does not rescue eye phenotypes. The lz mr1 eye remains the same in a p35 background (c, e); the lz L;pGMR-p35 eye is slightly "rougher" (i) than lz L (g). lz mr1 and lz L(d, h) exhibit minimal cell death in a pGMR-p35 background (f, j). k–n Eye and retinal phenotypes of ato 1/Df(3R)p13 and pGMR-p35/Y;ato 1/Df(3R)p13. SEMs reveal that the eye is a patch of pigment with few sensory hairs (k, m). Surface area measurements (n =4) reveal the pGMR-p35;ato pigment patch (m) is 25% larger than that in ato (k; n =4; measurements not shown). l, n Toluidine-blue-stained retinal sections of ato 1/Df(3R)p13 and pGMR-p35/Y;ato 1/Df(3R)p13 (black arrow shows antenna, white arrow shows pigment patch). Sections reveal a larger, denser patch in pGMR-p35/Y;ato 1/Df(3R)p13 (n; patch shown by arrowhead in both mutants)
Programmed cell death can be blocked by p35 expression
p35 functions as a broad-acting inhibitor of caspases and plays a role in interrupting apoptosis (Bump et al. 1995). To assess whether p35 could suppress lz phenotypes, the glass multimer repeat promoter (pGMR) was used to control p35 expression in eye discs. The GMR promoter drives expression in all cells in and behind the morphogenetic furrow in the developing eye (Hay et al. 1994). In lz mr1;pGMR-p35 discs, cell death could be blocked (Fig. 4f), but the lz mr1 eye phenotype persisted (Fig. 4e). In lz L;pGMR-p35, cell death was blocked (Fig. 4j), but the eye phenotype was altered slightly, giving it a "rougher" appearance indicative of ommatidia trying but failing to form (Fig. 4i).
These results suggest that cell death alone cannot account for eye defects in lz mutants, because cells that survive still fail to differentiate. At best, we observe minor amelioration of lz L;pGMR-p35 eyes, with changes in pigmentation (not shown) suggesting the presence of extra secondary or tertiary pigment cells. Similar observations are made in atonal (ato) mutants, where the fate of photoreceptor cell R8 is never established (Jarman et al. 1994; Dokucu et al. 1996) and only pigment cells are made (Fig. 4k–l). This may be considered as the "default" pathway of eye development. Apoptosis occurs posterior to the furrow in ato mutants (Jarman et al. 1994). We found that blocking apoptosis in ato mutants by pGMR-p35 expression resulted in a 25–30% increase in eye size (Fig. 4m; % increase based on surface eye measurements, not shown). The size increase is a result of extra secondary pigment cells that have taken the default pathway (Fig. 4n).
Cell death in D-Pax2 mutants
Since lz is known to play a part in the regulation of D-Pax2 in eye development, D-Pax2 mutants were assayed for cell death. D-Pax2 mutations result in moderate to severe eye phenotypes, where cone and pigment cell development is perturbed (Fig. 5a, b, d, e). Mutants show fenestrated membrane defects, resulting in photoreceptor cells intermingling with the first optic ganglia (Fig. 5e). Lens defects are also evident in D-Pax2 mutants (Fig. 5e), indicative of cone cell perturbation. However, some cone cells are still present in these mutants.

D-Pax2 mutants exhibit pigment and cone cell defects and also exhibit excess cell death in early development. a–c SEMs of spa P65 (a), spa pol (b) and pGMR-p35;spa pol (c). spa pol with pGMR-p35/+ is inset (c′). d–f Toluidine-blue-stained retinal sections. spaP65 has pigment cell defects with failure of the fenestrated membrane (d; arrow). spa pol (e) exhibits cone and pigment cell defects, the latter causing photoreceptors to be found in the lamina (la). pGMR-p35;spa pol (f) has excessive secondary (2°) pigment cells (arrowhead) and very few lenses. f′ A higher magnification view of the predominance of pigment cells (arrowhead) in the pGMR-p35;spa pol eye. g–i Acridine orange (AO) staining shows elevated, patterned cell death (arrowhead marks furrow) in spa P65 and spa pol(g, h). Hoechst/AO double staining allows unequivocal identification of cell nuclei and developing clusters revealing that apoptosis is about twelve to fifteen rows posterior to the furrow (h′). The pGMR-p35;spa pol eye disc exhibits very little cell death (i)
When assayed for cell death, all D-Pax2 mutants tested exhibited elevated levels of AO staining relative to a wild-type strain (Fig. 5g, h). Furthermore, abnormal cell death occurred in a synchronised band twelve to fifteen rows posterior to the furrow (Fig. 5h′); more posterior than the death observed in lz mutants (Fig. 2b, c, e, f). This result is indicative of a developmentally later onset of death occurring in D-Pax2 mutants than that in lz mutants. This is consistent with the role that lz plays in regulating D-Pax2.
When D-Pax2 mutants were crossed into a pGMR-p35 background, cell death was blocked (results shown for pGMR-p35;spapolonly; Fig. 5i). Interestingly, the block in apoptosis does not result in a rescued adult eye (Fig. 5c), but results in an eye lacking cone cells but having excess secondary pigment cells (Fig. 5f). This change in cell fate occurs when apoptosis is blocked and, among other possibilities, can be attributed to either autonomous cell fate transformation of cone cells into secondary pigment cells, or a disturbance in cell signalling mechanisms that induce secondary pigment cell development.
Discussion
Patterned programmed cell death results from mutations in lozenge and D-Pax2
A link between lz mutations and ectopic cell death in early eye development has clearly been established in this study, since cell death levels could be correlated with phenotypic severity in lz mutants (Fig. 1). The precise synchrony of cell death correlates with the expression pattern of lz and subsequent gene disruption (Crew et al. 1997; Flores et al. 1998).
D-Pax2 mutants exhibit a patterned wave of apoptosis occurring later in development than that in lz mutants (Fig. 5), consistent with the timing of D-Pax2 expression, which influences cone and primary pigment cell development (Fu and Noll 1997). In the absence of D-Pax2, cone and primary pigment cells continue to develop. Cells that die prematurely in D-Pax2 mutants appear to be unspecified cells that give rise to secondary and tertiary pigment cells.
lz has been shown to play a role in the direct regulation of D-Pax2 (Flores et al. 2000). However this study reveals that the causes and consequences of death are different in lz and D-Pax2 mutants. Hence, separate models for lz and D-pax2 control over death and the resulting phenotypes have been considered.
Explanations for ectopic cell death observed in early eye development
Many explanations have been proposed for ectopic apoptosis observed in mutants exhibiting eye defects. These include the failure of eye progenitor cell development as observed in eyeless mutants (Halder et al. 1995), a failure of fate determination as observed in yan and atonal mutants (Jarman et al. 1994; Rebay and Rubin 1995), or a failure to produce necessary survival signals such as in Ras pathway and EGFR signalling mutants (Bergmann et al. 1998; Domínguez et al. 1998; Kurada and White 1998; Baker and Yu 2001). Since lz is not expressed prior to furrow progression (Crew et al. 1997; Flores et al. 1998), either of the latter two explanations are likely to account for the death in lz mutants.
Instances in which cell death follows a failure of fate determination are well documented. Ectopic death observed in the posterior rim of yan eye discs has been attributed to cells choosing death as a "last resort" after a lengthy block of differentiation (Rebay and Rubin 1995). A similar pattern of death is observed in atonal mutants, where the fate of photoreceptor R8 is never established, causing ectopic death due to no other neuronal fate determination (Jarman et al. 1994; Dokucu et al. 1996).
In contrast, lz mutants exhibit a synchronous pattern of apoptosis occurring as a co-ordinated wave early in the differentiation process (Figs. 2a–f, 3b–i). The timing of death and the apical position of the pycnotic nuclei illustrates that cells are dying before or during their effort to establish a fate (Fig. 2g–j). Furthermore, the death is observed very early in lz null mutants, before lz-dependent cells have been added to their ommatidial clusters. This observation is not consistent with cells dying simply because they have failed to differentiate.
lz has dual roles in the developing eye
The early expression of lz in undifferentiated cells of the developing eye posterior to the furrow has been documented (Crew et al. 1997; Flores et al. 1998). These unfated, G1-arrested cells would normally be instructed to undergo cell death (reviewed in Baker 2001). However, in eye development, these cells must survive for several hours before other mechanisms that control fate determination, such as EGF receptor activation, lz control over genes including seven-up and Bar, and activation of the sevenless signalling cascade (Daga et al. 1996; Crew et al. 1997; Freeman 1997) can take over. During this time, the default cell death pathway must be repressed. The results of this study lead us to propose that the initial function of lz is to create a permissive environment, allowing cells to survive long enough to allow the R7 equivalence group cells to establish their fates. Thus lz has a role in cell survival.
Recently, lz has been implicated as being downstream of Yan in the Ras signalling pathway (Behan et al. 2002). Kurada and White (1998) showed that Pnt, a positive effector of the Ras pathway, downregulated hid expression. In contrast, the negative effector, Yan, upregulated hid thereby inducing cell death in embryos. They concluded that Ras, acting through the Map kinase pathway factors Pnt and Yan, induces differentiation and inhibits cell death. Since lz is proposed to be downstream of Yan, it is consistent that lz plays a role in cell survival, initially required to downregulate genes that activate cell death (Fig. 6a). The EGFR receptor has also been shown to be required for cell survival in the developing eye (Domínguez et al. 1998; Baker and Yu 2001, Yu et al. 2002). Interestingly, the removal of EGFR results in cell death just posterior to the furrow (Domínguez et al. 1998), similar to that observed in lz mutant eye discs. Given the timing of death in lz mutants, it is possible that lz acts downstream of the EGFR-Ras/MAP kinase signalling pathway, having a role in cell survival.

Models for the roles of lz and D-Pax2 in control of cell death and fate determination; based on previous reviews (Wolff et al. 1997; Freeman 1997, 1998) and on the primary work of Crew et al. (1997), Fu and Noll (1997) and Miller and Cagan (1998). Further studies are needed to test these models. a Eye discs exhibit a developmental time-line revealing progressive cell recruitment. The anterior (top) is least mature, growing through proliferation. Furrow (mf) progression induces synchronous G1 arrest. Events within the furrow induce photoreceptor R8 (blue cells; row 1). Rows represent steps in cell recruitment: row 2, recruitment of R2/5, row 3, R3/4. Lz expression is initiated in undifferentiated cells (yellow). The disc is then repopulated through mitosis (red). Recruitment continues with lz-expressing R1/6 and R7 (yellow). Boxes parallel the timing of fate determination. Lz initially contributes to repression of genes in the cell death pathway. Upon R1/6 recruitment, Lz represses svp and enhances Bar expression. Lz positively regulates prospero in R7 and cone cells. As cone and pigment cells join, Lz regulates D-Pax2 expression, which regulates cut and Bar (Fu and Noll 1997). In lz mutants, the cell death pathway is de-repressed, leading to premature apoptosis (skull and crossbones). Apoptotic death is blocked by p35 expression in lz mutants, but other regulatory functions that Lz serves are lacking. b In pupae, cell recruitment adds primary pigment cells (1°), then secondary and tertiary pigment cells (2°/3°). D-Pax2 is normally expressed in cone and 1° pigment cells (yellow). Cell-cell signals are provided first by these cells (black arrows), then by inter-ommatidial cells communicating within themselves (blue arrows). These signals regulate the activation of the EGF receptor on the inter-ommatidial cells, thus dictating how cells will develop (Freeman 1997; Miller and Cagan 1998). In spa pol, D-Pax2 absence (white) disrupts control of signalling mechanisms causing inter-ommatidial precursors to initiate a death program. Although 2° and 3° pigment cells are absent in spa pol mutants, cone and 1° pigment cells develop. In pGMR-p35;spa pol cell death is blocked, causing inter-ommatidial precursors to begin a self determination similar to that observed in atonal mutants where only pigment cells differentiate (Jarman et al. 1994). In this case, the inter-ommatidial precursor cell's own EGF receptor activation is the cue to develop (blue arrows). The signal is also presented to cone and 1° pigment cells (black arrows), which are developmentally delayed. These cells are subverted into 2° pigment cell differentiation. Thus, adult eyes lack lenses (normally formed by cone and 1° pigment cells) and are also pigmented by 2° pigment cells
Further evidence supporting the hypothesis that lz plays a role in cell survival in Drosophila eye development comes from the known function of another Runt domain protein in humans. AML1, like Lz, is a member of the RUNX (Runt/AML1) family of transcription factors and shares 71% identity to Lz within the Runt domain (Daga et al. 1996). The AML1 gene is involved in several types of chromosomal translocations associated with human acute myeloid leukemia. Interestingly, AML1, when overexpressed in T cell hybridoma lines, functions antagonistically against T cell receptor- (TCR) mediated apoptosis, by down regulation of a specific factor required for apoptotic death (Fujii et al. 1998).
The functional relevance of the lz gene in cell survival has been shown in this study. The timing of Lz expression and the timing of normal apoptotic waves in pupal development are also consistent with these results. Lz expression ceases in the pupal retina by 40 h after puparium formation (Flores et al. 1998). Slightly later, the pupal retina shows abundant apoptosis, removing unfated cells in a co-ordinated process (Wolff and Ready 1993).
Although the caspase inhibitor, p35, can block apoptosis in lz eye discs (Fig. 6a), lz adult eye phenotypes persist in lz mutants expressing elevated levels of p35. This is not surprising, given the direct role of lz in regulating genes required for cell fate determination (Daga et al. 1996; Crew et al. 1997; Flores et al. 2000). A model is proposed for the dual roles of lz in eye development, firstly as a cell survival factor, and secondly as a factor involved in cell fate specification (Fig. 6a).
The role of D-Pax2 mutations in producing increased levels of cell death
This study has revealed that while D-Pax2 mutations cause ectopic cell death in third instar eye discs, the mechanism and timing of death is different to that in lz mutants. D-Pax2 is normally expressed in cone and primary pigment cells. When D-Pax2 expression is removed, these cell types still differentiate (Fig. 5; Fu and Noll 1997). The apoptosis that occurs early in D-pax2 mutants results in a lack of secondary (2°) and tertiary (3°) pigment cells. Surprisingly, blocking cell death with p35 leads to an eye lacking cone and primary (1°) pigment cells, but produces excess 2° and 3° pigment cells (Fig. 5). This could be the result of autonomous cone and 1° pigment cell transformation to 2° and 3° cell fates, or the result of defective EGFR signalling causing non-autonomous cell fate transformation.
In eye development, cell recruitment relies on sequential inhibition and activation of the EGF receptor through proteins including Argos and Spitz (Freeman 1997, 1998; Domínguez et al. 1998). Spitz signalling is provided by cone and 1° pigment cells to induce 2° and 3° pigment cell development. The absence of cone and 1° pigment cells removes the Spitz signal. Absence of Spitz signalling leads to the premature death of inter-ommatidial precursors (Miller and Cagan 1998). Interestingly, 2° pigment cells can develop in the absence of photoreceptor and cone cells. Such a default program is evident in atonal mutants, where eyes lack photoreceptor and cone cells, but still generate pigment cells (Jarman et al. 1994). One possible explanation for the eye phenotype observed in pGMR-p35; spa pol flies is that rescued inter-ommatidial precursor cells could have the capacity to initiate the EGF receptor signalling mechanisms used to identify 2° pigment cells. These inter-ommatidial cells may then produce sufficiently high levels of signalling to overwhelm the developmental program, transforming available cells into 2° pigment cells. This model is consistent with the phenotype observed in pGMR-p35;spa pol flies (Fig. 5). The cartoon in Fig. 6b illustrates a model for this observed phenotype, though further studies will be needed to test this concept.
In conclusion, this study suggests Lz is an early survival signal in eye development, required to create a permissive environment where cell signalling events and fate determination can proceed. Lz may do this by direct or indirect repression of members of the cell death pathway. Ectopic cell death observed in lz mutants is not a consequence of failure of cells to differentiate. In contrast, defective lz expression causes de-repression of the cell death pathway, leading to premature death before other fates have had the opportunity to be established. In pupal development, lz expression ceases by 40 h after puparium formation (Flores et al. 1998). Slightly later, abundant apoptotic death is observed, removing unfated cells in a co-ordinated process. This observation is also consistent with the notion that lz plays a role in cell survival in the developing eye.
In D-Pax2 mutants, cells fail to correctly signal their neighbouring cells, leading to the premature death of neighbouring cells. Analysis of pGMR-p35; spa pol flies suggests that when cone and 1° pigment cell differentiation is delayed, cell fates can be transformed non-autonomously by signals provided by the developing 2° pigment cells or that the cells undergo autonomous cell fate transformation.
References
Abrams JM (1999) An emerging blueprint for apoptosis in Drosophila. Trends Cell Biol 9:435–440
Baker NE (2001) Cell proliferation, survival, and death in the Drosophila eye. Semin Cell Dev Biol 12:499–507
Baker NE, Yu SY (2001) The EGF receptor defines domains of cell cycle progression and survival to regulate cell number in the developing Drosophila eye. Cell 104:699–708
Bangs P, White K (2000) Regulation and execution of apoptosis during Drosophila development. Dev Dyn 218:68–79
Batterham P, Crew JR, Sokac AM, Andrews JR, Pasquini GM, Davies AG, Stocker RF, Pollock JA (1996) Genetic analysis of the lozenge gene complex in Drosophila melanogaster: adult visual system phenotypes. J Neurogenet 10:193–220
Baumann O, Walz B (1989) Topography of a Ca2+-sequestering endoplasmic reticulum in photoreceptors and pigmented glial cells in the compound eye of the honeybee drone. Cell Tissue Res 255:511–522
Beeson VS, Bender (1975) Phenogenetics of a suppressor and an enhancer gene of the lozenge34 k allele of Drosophila melanogaster. J Exp Zool 193:177–190
Behan K, Nichols CD, Cheung TL, Farlow A, Hogan BM, Batterham P, Pollock JA (2002) Yan regulates Lozenge during Drosophila eye development. Dev Genes Evol 212:267–276
Bergmann A, Agapite J, McCall K, Steller H (1998) The Drosophila gene hid is a direct molecular target of Ras-dependent survival signaling. Cell 95:331–341
Bonini NM, Fortini ME (1999) Surviving Drosophila eye development: integrating cell death with differentiation during formation of a neural structure. Bioessays 21:991–1003
Bump NJ, Hackett M, Hugunin M, Seshagiri S, Brady K, Chen P, Ferenz C, Franklin S, Ghayur T, Li P, et al (1995) Inhibition of ICE family proteases by baculovirus antiapoptotic protein p35. Science 269:1885–1888
Canon J, Banerjee U (2000) Runt and Lozenge function in Drosophila development. Semin Cell Dev Biol 11:327–336
Chen P, Nordstrom W, Gish B, Abrams JM (1996) grim, a novel cell death gene in Drosophila. Genes Dev 10:1773–1782
Crew JR, Batterham P, Pollock JA (1997) Developing compound eye in lozenge mutants of Drosophila: lozenge expression in the R7 equivalence group. Dev Genes Evol 206:481–493
Daga A, Karlovich CA, Dumstrei K, Banerjee U (1996) Patterning of cells in the Drosophila eye by Lozenge, which shares homologous domains with AML1. Genes Dev 10:1194–1205
Dokucu ME, Zipursky SL, Cagan RL (1996) Atonal, rough and the resolution of proneural clusters in the developing Drosophila retina. Development 122:4139–4147
Domínguez M, Wasserman JD, Freeman M (1998) Multiple functions of the EGF receptor in Drosophila eye development. Curr Biol 8:1039–1048
Finley RL Jr, Thomas BJ, Zipursky SL, Brent R (1996) Isolation of Drosophila cyclin D, a protein expressed in the morphogenetic furrow before entry into S phase. Proc Natl Acad Sci USA 93:3011–3015
Flores GV, Daga A, Kalhor HR, Banerjee U (1998) Lozenge is expressed in pluripotent precursor cells and patterns multiple cell types in the Drosophila eye through the control of cell-specific transcription factors. Development 125:3681–3687
Flores GV, Duan H, Yan H, Nagaraj R, Fu W, et al (2000) Combinatorial signaling in the specification of unique cell fates. Cell 103:75–85
Freeman M (1997) Cell determination strategies in the Drosophila eye. Development 124:261–270
Freeman M (1998) Complexity of EGF receptor signalling revealed in Drosophila. Curr Opin Genet Dev 8:407–411
Fu W, Noll M (1997) The Pax2 homolog sparkling is required for development of cone and pigment cells in the Drosophila eye. Genes Dev 11:2066–2078
Fu W, Duan H, Frei E, Noll M (1998) shaven and sparkling are mutations in separate enhancers of the Drosophila Pax2 homolog. Development 125:2943–2950
Fujii M, Hayashi K, Niki M, Chiba N, Meguro K, Endo K, Kameoka J, Ito S, Abe K, Watanabe T, Satake M (1998) Overexpression of AML1 renders a T hybridoma resistant to T cell receptor-mediated apoptosis. Oncogene 17:1813–1820
Gold R, Schmied M, Giegerich G, Breitschopf H, Hartung HP, Toyka KV, Lassman H (1994) Differentiation between cellular apoptosis and necrosis by the combined use of in situ tailing and nick translation techniques. Lab Invest 71(2):219–225
Grether ME, Abrams JM, Agapite J, White K, Steller H (1995) The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev 9:1694–1708
Halder G, Callaerts P, Gehring WJ (1995) Induction of ectopic eyes by targeted expression of the eyeless gene in Drosophila. Science 267:1788–1792
Hay BA, Wolff T, Rubin GM (1994) Expression of baculovirus P35 prevents cell death in Drosophila. Development 120:2121–2129
Jarman AP, Grell EH, Ackerman L, Jan LY, Jan YN (1994) Atonal is the proneural gene for Drosophila photoreceptors. Nature 369:398–400
Kurada P, White K (1998) Ras promotes cell survival in Drosophila by downregulating hid expression. Cell 95:319–329
Lindsley DL, Zimm GG (1992) The genome of Drosophila melanogaster. Academic Press, San Diego
McCall K, Steller H (1997) Facing death in the fly: genetic analysis of apoptosis in Drosophila. Trends Genet 13:222–226
McNeill H, Downward J (1999) Apoptosis: Ras to the rescue in the fly eye. Curr Biol 9:R176–179
Meier P, Evan G (1998) Dying like flies. Cell 95:295–298
Miller DT, Cagan RL (1998) Local induction of patterning and programmed cell death in the developing Drosophila retina. Development 125:2327–2335
O'Neill EM, Rebay I, Tijan R, Rubin GM (1994) The activities of two Ets-related transcription factors required for Drosophila eye development as modulated by the Ras/MAPK pathway. Cell 78:137–147
Pollock JA, Ellisman MH, Benzer S (1990) Subcellular localisation of transcripts in Drosophila photoreceptor neurons: chaoptic mutants have an aberrant distribution. Genes Dev 4:806–821
Rabizadeh S, LaCount DJ, Friesen PD, Bredesen DE (1993) Expression of the baculovirus p35 gene inhibits mammalian neural cell death. J Neurochem 61:2318–2321
Ready DF, Hanson TE, Benzer S (1976) Development of the Drosophila retina, a neurocrystalline lattice. Dev Biol 53:217–240
Rebay I, Rubin GM (1995) Yan functions as a general inhibitor of differentiation and is negatively regulated by activation of the Ras1/MAPK pathway. Cell 81:857–866
Reja V, Goodchild AK, Phillips JK, Pilowsky PM (2002) Tyrosine hydroxylase gene expression in ventrolateral medulla oblongata of WKY and SHR: a quantitative real-time polymerase chain reaction study. Autonom Neurosci Basic Clin 98:79–84
Rusconi JC, Hays R, Cagan RL (2000) Programmed cell death and patterning in Drosophila. Cell Death Differ 7:1063–1070
Spreij TE (1971) Cell death during development of the imaginal discs of Calliphora erythrocephala. J Neth Zool 3:221–264
Tautz D, Pfeifle C (1989) A non-radioactive in situ hybridization method for the localisation of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma 98:81–85
Tomlinson A, Ready DF (1987) Neuronal differentiation in the Drosophila ommatidium. Dev Biol 120:366–376
White K, Grether ME, Abrams JM, Young L, Farrell K, Steller H (1994) Genetic control of programmed cell death in Drosophila. Science 264:677–683
White K, Tahaoglu E, Steller H (1996) Cell killing by the Drosophila gene reaper. Science 271:805–807
Wolff T, Ready DF (1991) Cell death in normal and rough eye mutants of Drosophila. Development 113:825–839
Wolff T, Martin K, Rubin GM, Zipursky SL (1997) The development of the Drosophila visual system. In: Cowan WM, Jessell TM, Zipursky SL (eds) Molecular and cellular approaches to neural development. Oxford University Press, Oxford, pp 474–508
Xu C, Kauffmann RC, Zhang J, Kladny S, Carthew RW (2000) Overlapping activators and repressors delimit transcriptional response to receptor tyrosine kinase signals in the Drosophila eye. Cell 103:87–97
Yu SY, Yoo SJ, Yang L, Zapata C, Srinivasan A, Hay BA, Baker NE (2002) A pathway of signals regulating effector and initiator caspases in the developing Drosophila eye. Development 129:3269–3278
Acknowledgements
We appreciate the thoughtful comments provided by the anonymous referees. We also extend our gratitude to Dr. M. Green who generously provided insight and guidance. We thank the Drosophila Stock Center at the University of Indiana, Bloomington, the Mid-America Stock Center, Bowling Green, and Drs. R. Stocker and M. Green for many of the fly strains used in this study. Dr. H. Steller provided a rpr clone. Dr. Rubin provided strains expressing baculovirus p35. Elav antibody developed by G. Rubin was obtained from the Developmental Studies Hybridoma Bank maintained by the University of Iowa, Department of Biological Sciences, Iowa City, IA. Technical assistance was provided by J. Suhan for the thin tissue sectioning and aspects of electron microscopy. We are grateful to Dr. U. Banerjee for valuable discussion and for sharing fly strains and unpublished results. R. Selvaraju provided thoughtful commentary on the manuscript. This work was supported in part by Duquesne University of the Holy Ghost and the following grants: NIH Pre-doctoral fellowship 5T32GM08067–100031 to J.R.C.; to J.A.P SURG/HHMI Awards to undergraduate researchers at Carnegie Mellon University, NIH grant EY09093, NSF/STC grant BIR-8920118, March of Dimes Birth Defects Foundation FY93–1010; Carnegie Mellon University Faculty Development Awards; Samuel and Emma Winters Foundation; and international collaborative support from the NSF INT-9605205 and the University of Melbourne.
Author information
Authors and Affiliations
Corresponding author
Additional information
K.J. Behan, J.R. Crew, and J.A. Pollock conducted aspects of this work in the Department of Biological Sciences, Carnegie Mellon University. Pittsburgh, PA
Edited by P. Simpson
Rights and permissions
About this article
Cite this article
Siddall, N.A., Behan, K.J., Crew, J.R. et al. Mutations in lozenge and D-Pax2 invoke ectopic patterned cell death in the developing Drosophila eye using distinct mechanisms. Dev Genes Evol 213, 107–119 (2003). https://doi.org/10.1007/s00427-003-0295-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00427-003-0295-y