
Abstract
Calcium homeostasis in the cardiomyocyte is critical to the regulation of normal cardiac function. Abnormal calcium dynamics such as altered uptake by the sarcoplasmic reticulum (SR) Ca2+-ATPase and increased diastolic SR calcium leak are involved in the development of maladaptive cardiac remodeling under pathological conditions. Ca2+/calmodulin-dependent protein kinase II-δ (CaMKIIδ) is a well-recognized key molecule in calcium dysregulation in cardiomyocytes. Elevated cellular stress is known as a common feature during pathological remodeling, and c-jun N-terminal kinase (JNK) is an important stress kinase that is activated in response to intrinsic and extrinsic stress stimuli. Our lab recently identified specific actions of JNK isoform 2 (JNK2) in CaMKIIδ expression, activation, and CaMKIIδ-dependent SR Ca2+ mishandling in the stressed heart. This review focuses on the current understanding of cardiac SR calcium handling under physiological and pathological conditions as well as the newly identified contribution of the stress kinase JNK2 in CaMKIIδ-dependent SR Ca2+ abnormal mishandling. The new findings identifying dual roles of JNK2 in CaMKIIδ expression and activation are also discussed in this review.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the mammalian heart, calcium (Ca2+) is an essential regulator of electrical signals, contractile function, and excitation–contraction coupling (ECC) during each heartbeat and also plays an important role in the cellular signal transduction pathways that control myocyte survival and growth [17, 42, 116]. Under physiological conditions, the heart beats more than two billion times during an average human lifespan to supply blood to the body. With increasing age and abnormal conditions such as heart failure (HF) and excessive alcohol intake, impaired Ca2+ homeostasis causes myocardial molecular remodeling including aberrant gene expression, myocyte death, contractile dysfunction, and arrhythmias [2, 111, 150]. Ca2+/calmodulin-dependent protein kinase II (CaMKII) is a multifunctional signaling molecule that plays a central role in this impaired Ca2+ homeostasis promoting maladaptive cardiac remodeling and arrhythmias [2, 6, 91, 155,156,157].
Under pathological conditions, intrinsic cellular stresses caused by a number of stimuli including oxidative stress, ischemia, and inflammation are markedly enhanced. These stressors are also well-established risk factors for the development of cardiovascular diseases [12, 14, 62, 64, 103]. The c-jun N-terminal kinase (JNK), a member of the mitogen-activated protein kinase (MAPK) family, is activated in response to various stress challenges [29, 66, 112, 115]. In fact, JNK activation has been observed with aging, excessive binge alcohol intake-triggered “Holiday Heart Syndrome,” and with cardiovascular diseases such as ischemic myocardial infarction (MI) and HF [29, 63, 66, 80, 108, 112, 115, 136, 144]. Our lab recently discovered a novel sub-cellular mechanism describing a pathogenic kinase-to-kinase crosstalk between JNK2 kinase and the “pro-arrhythmic kinase” CaMKIIδ in the governance of intercellular Ca2+ signaling and consequently Ca2+-mediated arrhythmias.
A JNK kinase was first discovered in the early 1990s by Kyriakis and Avruch and reported as a novel protein called pp54 MAP-2 kinase, which is activated by duel phosphorylation of the amino acid residues of serine-183 and threonine-185 [74, 75]. Later, two isoforms were identified with molecular weights of 46 and 56 kDa and were named JNK1 and JNK2, respectively [58]. It was then revealed that these JNK kinases could be activated by various extracellular stimuli. Because JNK contains the threonine–tyrosine phosphorylation motif (TPY), it was thus characterized as a member of the MAPK family. Next, JNK3 was discovered in 1995 as the third member of this MAPK subfamily and is mainly expressed in neurons [67, 95, 99, 115]. In the heart, JNK1 and JNK2 are the major isoforms, while JNK3 is expressed at a much lower level [73]. In this review, we will discuss the new findings identifying dual roles of JNK2 in CaMKIIδ expression and activation and the current understanding of the crucial role of the stress kinase JNK2 isoform in CaMKIIδ-dependent aberrant sarcoplasmic reticulum (SR) Ca2+ handling in stressed hearts.
Physiological calcium dynamics in the heart
Ca2+ is an important cation in the conversion of an electrical signal to mechanical function in the heart from beat to beat [1, 88, 93, 118, 126]. The voltage-gated L-type Ca2+ channels (LTCCs) located in the plasma membrane are activated by depolarization of the myocyte membrane, which leads to a small amount of inward Ca2+ flux (ICa) [44, 50, 81, 117, 123, 130]. This Ca2+ entry through LTCCs triggers large quantities of Ca2+ to be released from the SR via cardiac ryanodine receptor type-2 (RyR2; also called Ca2+-triggered SR Ca2+ release channels) [9, 84, 93]. The plasma membrane and SR are coupled to allow this Ca2+-induced Ca2+ release (CICR), which occurs locally within the clusters of RyR2 channels on the SR membrane that are in close proximity to LTCCs [59, 77]. CICR is further facilitated by dyads, which are the structures consisting of terminal cisternae of SR, composed of RyR2 channels, paired with transverse tubules (T-tubules), and LTCCs [139]. When an action potential arrives at the T-tubule, Ca2+ influx via LTCCs activates RyR2 channels on the cytosolic side of the SR allowing the occurrence of CICR, which activates neighboring RyR2 channels within the same dyad, resulting in a rapid increase in cytosolic Ca2+ [45, 76]. CICR is also the trigger for Ca2+-troponin C binding, leading to myofilament activation and cardiac muscle contraction [86, 89].
During cardiac muscle relaxation, LTCCs close and terminate the influx of Ca2+ and cytosolic Ca2+ is removed through the sodium–calcium exchanger (NCX) to the extracellular space and pumped back to the SR through the cardiac sarcoplasmic reticulum Ca2+-ATPase (SERCA2), while another small portion of Ca2+ is taken up by mitochondria via mitochondrial Ca2+ uniporters as well as a small Ca2+ efflux via the plasma membrane Ca2+-ATPase (PMCA) [10, 17, 34, 50, 107]. Normal contraction of the heart requires high Ca2+ levels in systole and low levels in diastole [35, 69]. Therefore, SR Ca2+ release via RyR2 channels and reuptake via the predominant Ca2+ pump SERCA2a isoform (SERCA2a), and to a much lesser extent SERCA2b isoform, critically mediate the cytoplasmic Ca2+ concentration, which is essential in cardiac contraction and relaxation of each heartbeat [1, 118].
Functional impacts of pathological SR Ca2+ mishandling
Given the tightly regulated role of Ca2+ in ECC, even a small amount of aberrant Ca2+ release resulting from slowly developing pathological changes in the intracellular Ca2+ homeostasis can potentially have escalating negative consequences for the myocyte and ultimately the whole heart. Under pathological conditions including HF, ischemia–reperfusion (IR) injury, post-MI, atrial fibrillation (AF), and ventricular arrhythmias, abnormal SR Ca2+ dynamics result in electrical and mechanical dysfunctions and myocardial maladaptive function (Fig. 1) [2, 18, 21, 22, 30, 68, 82, 145, 147].
For instance, HF is a common disorder in which the cardiac output does not meet the needs of the body resulting from dysfunctional contractility, impaired electrical conduction, and abnormal energy metabolism [22, 30]. During the SR Ca2+ cycling, decreased SR Ca2+ refill via reduced Ca2+ uptake by SERCA2a leads to a reduced Ca2+ transient amplitude and consequently decreased cardiac contractility as seen in the failing heart [57, 83, 125, 142]. In the diastolic phase, SR Ca2+ release normally shuts off almost completely (∼99%). However, increased diastolic RyR2 channel activity could be responsible for increased diastolic SR Ca2+ leak and reduced systolic fractional Ca2+ release for a given L-type voltage-gated Ca2+ current (Ica) as the release trigger [11, 19, 121]. The increased diastolic SR Ca2+ leakage along with an impaired SR Ca2+ uptake in HF slows down the intracellular Ca2+ decline and then elevates the amount of diastolic intracellular Ca2+ concentration, which leads to increased sodium (Na+) influx via NCX for removing the elevated intracellular Ca2+ outside of the cell membrane. As a result, increased diastolic SR Ca2+ leak promotes aberrant Ca2+ events (Ca2+ sparks and waves) and the inward NCX current produces abnormal triggered activities such as delayed after-depolarizations (DADs) and initiates atrial arrhythmias such as atrial fibrillation (AF, the most common cardiac arrhythmia) and ventricular arrhythmias including ventricular tachycardia and ventricular fibrillation (a fatal type of cardiac arrhythmia) [9, 17, 19, 31, 81]. This abnormal SR Ca2+ handling also occurs in AF pathogenesis as discussed in detail below.
Ca2+/calmodulin-dependent protein kinase II in the pathological SR Ca2+ mishandling
One of the hallmarks of a diseased heart is that altered protein phosphorylation critically contributes to ion transporter and channel dysfunctions, which leads to the disruption of SR Ca2+ dynamics. CaMKII is a well-recognized pro-arrhythmic kinase, promoting abnormal SR Ca2+ dynamics via phosphorylation of Ca2+ handling proteins in the heart. There are four highly conserved isoforms of CaMKII (α, β, γ, and δ) widely expressing in the body, while the δ isoform is predominantly expressed in the heart. Extensive studies demonstrate that activated CaMKIIδ is critically involved in phosphorylation of RyR2 at the site of Ser2815, resulting in sensitized RyR2 channels and profoundly increased diastolic SR Ca2+ leak that in turn promotes triggered activities and arrhythmia initiation in pathologically altered ventricles in HF [2, 6, 51, 56, 87, 91, 111, 127, 150, 155, 156, 158]. Although protein kinase A (PKA) has also been shown to phosphorylate RyR2 channels, inconsistent findings regarding the arrhythmic effect of PKA hyperphosphorylation of RyR2 at Ser2809 in HF were reported [3, 7, 16, 85, 90, 110, 120, 135, 138, 140, 154], further suggesting the key role of CaMKIIδ in HF-evoked arrhythmias. In recent years, accumulating evidence suggests that CaMKIIδ-dependent RyR2 channel dysfunction also leads to SR Ca2+ mishandling and triggered activities (delayed afterdepolarizations (DADs)) in the atria of chronic AF patients and post-operative AF patients [55, 131]. For instance, activated CaMKII was found to increase both arrhythmic Ca2+ activities and profibrotic activity-caused structural remodeling in chronic AF patients associated with HF [28, 96]. In post-operative AF patients (with no history of AF prior to the open-chest surgery), activation of the inflammasome signaling protein, NACHT, LRR, and PYD domain containing protein 3 (NLRP3), was found to augment the CaMKIIδ-dependent RyR2-hyperphosphorylation and arrhythmic Ca2+ activities [25, 26, 100]. Further, this CaMKIIδ-dependent SR Ca2+ mishandling was recently found to underlie AF pathogenesis in rabbit and mouse models of aging and holiday heart syndrome, in a tachy-pacing canine AF model, and in a spontaneous AF CREM mouse model [33, 79, 134, 145]. Similar to the regulatory actions of CaMKIIδ in the RyR2 activity, CaMKIIδ is known to enhance the Ca2+ binding affinity of SERCA2a by phosphorylating phospholamban (PLB) at the threonine-17 site (PLB17) to release the inhibitory PLB from SERCA2a, which enhances SERCA2a Ca2+ affinity and stimulates SR Ca2+ uptake [124]. The inhibitory effect of PLB on the SERCA2a activity contributes to, at least in part, reduced SR Ca2+ uptake in failing hearts [27]. In addition to the critical role of activated CaMKIIδ in SR Ca2+ handling, CaMKIIδ is also known to regulate other ion channels such as Ca2+ [114, 137], Na+ [53, 132, 151], and K+ [98, 102] channels and NCX [143, 149]. For instance, CaMKIIδ-dependent phosphorylation of cardiac voltage-gated sodium channel isoform 1.5 (NaV1.5) enhances a late depolarizing current (INa. Late), which leads to prolonged action potentials (APs) and disrupted Ca2+ handling and promotes arrhythmogenic DADs [48, 71]. In addition to Na+ channels, it has been suggested that CaMKIIδ activation regulates potassium Kv channels and reduces Ito and IK1 currents, which prolong APs and increase Ca2+-triggered repolarization, and ultimately enhance arrhythmogenicity [78, 133, 141]. Thus, CaMKIIδ is an important arrhythmic kinase playing a crucial role in the cardiac Ca2+ homeostasis and ECC in pathologically remodeled hearts (Fig. 1). Because CaMKIIδ is the predominant pro-arrhythmic isoform in the heart, CaMKIIδ inhibition has been considered a potential therapeutic approach to treat heart diseases [4, 5, 43, 72, 113]. Thus, understanding the underlying mechanisms of CaMKIIδ activation is of vital importance.

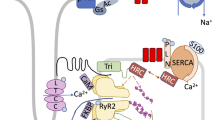
A representation of normal Ca2+ cycling in cardiomyocytes and stress kinase JNK2-mediated CaMKIIδ-dependent SR Ca2+ mishandling and maladaptive cardiac dysfunction. Physiological and pathological regulation of Ca2+ cycling in cardiomyocytes and activated CaMKIIδ-dependent dysregulation of RyR2 as well as other ion channels (i.e., NaV1.5 and KV), which lead to enhanced diastolic SR Ca2+ leak, triggered arrhythmic Ca2+ activates (sparks and waves), DADs, and cardiac dysfunction. A JNK2-driven CaMKIIδ-dependent diastolic leak–uptake relationship also enhances triggered arrhythmic activities. JNK2 c-jun N-terminal kinase, CaMKIIδ Ca2+/calmodulin-kinase type-II delta isoform, NaV1.5a voltage-gated sodium channel isoform 1.5-alpha, LTCC L-type Ca2+ channels, OX oxidation, SR sarcoplasmic reticulum, PLB phospholamban, SERCA2 SR Ca2+-ATPase, DADs delayed afterdepolarizations, P phosphorylation, RyR2 ryanodine receptor type-2, NCX sodium-calcium exchanger
A novel finding of stress kinase JNK2-regulated CaMKIIδ activation
In the past decades, significant progress has been made regarding the underlying mechanisms of CaMKIIδ activation under pathological conditions [39,40,41, 52, 155]. CaMKIIδ is a serine/threonine kinase with an increased activity when the site of threonine-287 is phosphorylated that leads to increased binding of its regulatory region to Ca2+/calmodulin, exposing CaMKIIδ to the kinase substrate and ATP binding sites and allowing phosphorylation of the target proteins by CaMKIIδ. Several elegant studies [39,40,41, 52] revealed different underlying mechanisms of post-translational modification including oxidization at the Met281/Met282 sites, S-nitrosylation on Cys290, and O-GlcNAcylation on Ser279, which all can lead to sustained activation of CaMKIIδ under various pathological conditions (Fig. 2). On the other hand, protein phosphatases such as PP1 are also important in maintaining the activation status of CaMKIIδ in HF and AF [23, 36, 60, 101, 152]. In patients with myocardial infarction and angina, increased protein phosphatase 1 (PP1) and reduced endogenous PP1 inhibitory protein inhibitor-1 (I-1) were associated with reduced CaMKII activity via dephosphorylation of the autophosphorylation site Thr287 of CaMKIIδ, while I-1 KO mice also showed reduced CaMKIIδ activity in the heart [101, 152]. In chronic AF, hyperphosphorylated I-1 suppresses the PP1 activity to consequently sustain the activation of CaMKIIδ [36, 54, 92]. However, reduced I-1 was also found in the failing heart, but it was associated with increased CaMKII activity due to the exchange protein activated by cAMP (EPAC)-augmented CaMKII activation [101]. Moreover, activation of CaMKIIδ could be sustained by reactive oxygen species (ROS) via inactivating phosphatases to reduce the protein phosphatase-regulated dephosphorylation of CaMKIIδ [5, 101]. Clearly, this kinase–phosphatase relationship is complex in the diseased heart and further investigation is needed. Nevertheless, all of the current findings emphasize the clinical significance of exploring effective approaches to inhibit CaMKIIδ activity as potential therapeutic strategies to prevent and/or treat cardiac diseases and arrhythmias.

A schematic outline of underlying mechanisms of CaMKIIδ activation and its functional consequences. Stressors (i.e., aging, alcohol, HF, and MI) lead to the activation of signal transduction pathways and the JNK kinase family (JNKs), which enhance CaMKIIδ activity through post-translational modifications including S-nitrosylation, O-GlcNacylation, oxidation, or direct phosphorylation on the regulatory domain of CaMKIIδ. Activated CaMKIIδ phosphorylates its downstream protein targets (i.e., RyR2, LTCC, and PLB) that are central to calcium homeostasis in the cardiomyocyte and in abnormal calcium dynamics in the development of maladaptive cardiac remodeling under pathological conditions (i.e., arrhythmia and HF). Auto-P autophosphorylation, Ca2+/CaM calmodulin, CVDs cardiovascular diseases, HF heart failure, LTCC L-type Ca2+ channels, MI myocardial infarction, NO nitric oxide, O-GlcNA O-linked-N-acetylglucosaminylation (O-GlcNAcylation), PLB phospholamban, RNS reactive nitrogen species, ROS reactive oxygen species, RyR2 ryanodine receptor type-2, S-Nitro s-nitrosylation.
Recently, our lab reported for the first time that the stress–response kinase JNK2 drives a kinase-to-kinase crosstalk as a previously unrecognized molecular mechanism of CaMKIIδ activation in both aging and binge alcohol-exposed holiday heart syndrome human and animal models [145, 147]. Notably, elevated cellular stress is a common feature of the heart under extrinsic stimuli or during pathological remodeling. JNK is a well-characterized kinase that is activated in response to intrinsic and extrinsic stress stimuli, and then modulates cellular functions including Ca2+ mishandling, cell death, and survival [29, 67]. JNK1 activation has been observed in various cardiovascular diseases including IR injury, MI, and HF, which frequently occur together in the aging population [15, 49, 66, 112, 115, 144]. However, the function of JNK2 in the heart has received less attention. Although not all hearts will experience a particular stress, all hearts will inevitably age. The aged heart is also known to be more susceptible to the stresses it may encounter [64]. We found that JNK2, but not JNK1, is significantly activated in both aged human and animal atria, while the levels of total JNK1 and JNK2 proteins were unchanged [144, 145]. Moreover, we found that activated atrial JNK2 is a consistent feature of aged atrium among different species (humans, rabbits, and mice) [144,145,146,147]. Furthermore, our recent unpublished data indicates that JNK2 also exhibits elevated activity in aged ventricles. Functionally, we revealed that this age-induced JNK2 activation directly phosphorylates CaMKIIδ to enhance its kinase activity and drive pathology [25, 113]. Intriguingly, we also discovered that JNK2 and CaMKIIδ proteins are tethered with each other and JNK2 increases phosphorylation of CaMKIIδ at the autophosphorylation site Thr287. Since protein phosphatase PP1 is also known to target this Thr287 site to dephosphorylate CaMKII [101, 129, 152], it could be a counterpoint to JNK2 in sustaining the phosphorylation status of CaMKII under stressed conditions. It is clear that this is worthy of further investigation. Accumulating evidence suggests that activation of the stress–response kinase JNK2 represents a common feature in many organs with either acute or chronic alcohol exposure, which contributes to alcohol-caused cell death and tissue injury [8, 37, 38, 104, 148]. Our lab further detailed a previously unknown link between binge alcohol exposure, JNK2 activation, enhancement of CaMKIIδ activity, and atrial arrhythmogenicity in humans and animal models of “holiday heart syndrome” [145]. Note that alcohol can increase ROS production [105] and elevated ROS does promote CaMKIIδ activation by oxidizing CaMKIIδ’s Met280 and Met281 sites, creating a dynamic methionine oxidation pathway for calcium-independent activation of CaMKIIδ [39]. However, our studies demonstrate that the JNK2-specific regulation of CaMKIIδ activation is independent of either intracellular Ca2+ concentration or oxidative stress [145, 147]. Therefore, JNK2 has a specific action in the CaMKIIδ activation in the stressed heart (Fig. 2).
Next, we found that JNK2-specific CaMKIIδ activation results in CaMKIIδ-dependent phosphorylation of RyR2815 and PLB17 in both aged and binge alcohol-exposed hearts and the functional consequence of this JNK2-specific regulation is enhanced arrhythmogenic diastolic SR Ca2+ activities and AF pathogenesis. Specifically, JNK2 increases diastolic SR Ca2+ leak via CaMKIIδ-dependent phosphorylation of RyR2, which sensitizes RyR2 channels, triggers aberrant Ca2+ waves, prolongs the intracellular Ca2+ decay time constant, enhances spatiotemporal heterogeneity of Ca2+ and electrical impulses, and augments AF susceptibility. Studies have also shown that an altered cellular redox balance towards a more oxidized state can also lead to oxidative modifications of RyR2, which promotes diastolic Ca2+ leak, arrhythmogenesis, and contractile dysfunction [13, 19, 20, 122, 160]. While we found JNK2-activated CaMKIIδ is independent of ROS, inhibition of either JNK2 or CaMKIIδ eliminates arrhythmogenic activities including enhanced diastolic leak, aberrant Ca2+ waves in myocytes, and enhanced arrhythmic susceptibility in intact heart and live animals. Therefore, our findings demonstrate that JNK2 acts as a key pathological node that transduces different stress stimuli and directly activates CaMKIIδ, which promotes SR Ca2+ mishandling in the heart and enhances arrhythmic susceptibility. The inter-relationship between ROS and JNK2 in CaMKIIδ activation remains to be determined. Overall, hyperactivation of CaMKIIδ under a stressed and/or diseased state leads to maladaptive cardiac remodeling including channel dysfunction, impaired Ca2+ homeostasis, and contractile dysfunction resulting in deteriorated cardiac function and increased risk of arrhythmias as summarized in Fig. 1. Our recent findings are significant because these JNK2-specific actions on CaMKIIδ activity and SR Ca2+ mishandling shed new light on modulating JNK2 as a new strategy to target CaMKIIδ activity for preventing and treating arrhythmias. It is clear that further investigation is needed to understand the potential functional roles of JNK2 on other ion channels and the inter-relationship between JNK2 and other kinases or protein phosphatases.
Transcriptional regulation of JNK2 in CaMKIIδ expression
Although CaMKIIδ is essential in regulating a large number of cellular substrates including ion channels, pumps, transporters, and transcription factors [113], how the CaMKIIδ gene and protein expression is controlled remains surprisingly understudied. We recently revealed for the first time that JNK2 plays an essential role in CaMKIIδ expression at the transcriptional level under both physiological and pathological conditions [46] (Fig. 3).

JNK2 kinase is schematically shown as an essential transcriptional regulator of CaMKIIδ expression. The JNK2 downstream transcription factors c-jun and ATF2 both bind to the CaMKIIδ gene promoter, but c-jun is a key transcription factor regulating the basal level expression of CaMKIIδ and a crucial transcriptional enhancer of CaMKIIδ expression in response to certain stress stimuli. ATF2 activating transcription factor 2
An extensive number of studies demonstrate that JNK1 activation is critically involved in the preservation of cardiac function and in promoting apoptosis after myocardial IR, MI, and HF via the regulation of signaling pathways that modulate gene expression [24, 29, 32, 70, 94, 106, 109, 115, 119, 159]. However, the function of cardiac JNK2, one of the two major cardiac isoforms, has surprisingly received less attention. It is known that JNKs directly regulate these cellular processes via direct phosphorylation of downstream targets and/or indirectly regulate gene expression via downstream transcription factors including c-jun and activating transcription factor 2 (ATF2), forming the activator protein-1 (AP-1) complex [46, 65, 97, 128, 146, 153]. The AP-1 complex is composed of homodimers of c-jun or heterodimers of c-jun/ATF2 or other combinations of transcription factors, which induce target gene expression by binding the AP-1 consensus site(s) in the promoter region of the gene or dissociating from the promoter region to upregulate or suppress the specific gene expression [47]. In our recent studies [46], we discovered that the JNK2 downstream transcription factor c-jun and ATF2 both bind to the CaMKIIδ gene promoter and upregulate CaMKIIδ expression. We further discovered that c-jun is surprisingly a key transcription factor for the basal level expression of CaMKIIδ mRNAs and proteins. This was evidenced by the suppression of CaMKIIδ promoter baseline activity when the c-jun binding consensus sequence was mutated. Moreover, robustly activated JNK2, mimicking a stressed condition, significantly increases the binding of c-jun, but not ATF2, to the CaMKIIδ promoter, while JNK2 inhibition alleviated this enhanced c-jun binding. In addition, the JNK2-specifiic action in c-jun-regulated CaMKIIδ promoter activity was supported by the suppressed CaMKIIδ promoter activity from either JNK2 or c-jun siRNA knockdown. Until very recently, the underlying mechanism of transcriptional regulation of CaMKIIδ gene expression remained completely unknown. Our discovery of the isoform-specific action of JNK2 in CaMKIIδ expression ( 3) provided the first evidence suggesting that JNK2 is not only an essential regulator in CaMKIIδ expression under physiological conditions but also is a crucial transcriptional enhancer in response to certain stress stimuli.
Conclusions and future directions
Over the years, our understanding of the underlying mechanisms of physiological Ca2+ homeostasis in cardiomyocytes and disrupted Ca2+ dynamics under pathological conditions has been significantly advanced. However, it is still not completely understood how stress stimuli and stress–response kinase JNKs are involved in aging, alcohol, obesity, and diseased states associated with cardiac Ca2+ mishandling and what mechanisms prompt cardiac maladaptive molecular and electrophysiological remodeling and cardiac dysfunction. While numerous studies have significantly advanced our understanding of the key role of hyperactivation of CaMKIIδ in pathological cardiac remodeling and arrhythmia, many questions remain. Examples are the relationship between pathologically hyperactivated CaMKIIδ and stress-activated JNKs, the interaction between JNK2/ CaMKIIδ and their downstream targets (i.e., RyR2, LTCCs, and PLB), and how stress JNK2 signaling and/or CaMKIIδ interact with other pathological signaling pathways during the process of disease development. All of these questions merit further investigation.
Accumulating evidence suggests that suppression of CaMKIIδ function can mitigate arrhythmias and various heart diseases in animal models provoking a great deal of interest in the development of CaMKIIδ inhibitors as possible anti-arrhythmic therapeutic agents [2, 46, 61, 155, 158]. Although a variety of CaMKIIδ inhibitors are currently available for research, their off-target effects hinder their clinical applications [61]. Thus, additional upstream or downstream components of the CaMKIIδ signaling cascades are being considered for new therapeutic approaches. As demonstrated by our recent studies, JNK2 as a key regulator of the pro-arrhythmic CaMKIIδ sheds new light on the possibility of modulating JNK2 activity as an alternative approach to targeting CaMKIIδ activity. This may offer broader clinical applications for treatment of AF, HF, holiday heart syndrome, and potentially other cardiovascular diseases.
Data Availability
Not applicable.
References
Afzal N, Dhalla NS (1992) Differential changes in left and right ventricular SR calcium transport in congestive heart failure. Am J Physiol 262:H868–H874. https://doi.org/10.1152/ajpheart.1992.262.3.H868
Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM (2005) Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res 97:1314–1322. https://doi.org/10.1161/01.RES.0000194329.41863.89
Alvarado FJ, Chen X, Valdivia HH (2017) Ablation of the cardiac ryanodine receptor phospho-site Ser2808 does not alter the adrenergic response or the progression to heart failure in mice. Elimination of the genetic background as critical variable. J Mol Cell Cardiol 103:40–47. https://doi.org/10.1016/j.yjmcc.2017.01.001
Anderson ME (2005) Calmodulin kinase signaling in heart: an intriguing candidate target for therapy of myocardial dysfunction and arrhythmias. Pharmacol Ther 106:39–55. https://doi.org/10.1016/j.pharmthera.2004.11.002
Anderson ME (2011) Pathways for CaMKII activation in disease. Heart Rhythm 8:1501–1503. https://doi.org/10.1016/j.hrthm.2011.04.027
Anderson ME, Brown JH, Bers DM (2011) CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol 51:468–473. https://doi.org/10.1016/j.yjmcc.2011.01.012
Antos CL, Frey N, Marx SO, Reiken S, Gaburjakova M, Richardson JA, Marks AR, Olson EN (2001) Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase a. Circ Res 89:997–1004. https://doi.org/10.1161/hh2301.100003
Aroor AR, Shukla SD (2004) MAP kinase signaling in diverse effects of ethanol. Life sciences 74:2339–2364
Ather S, Respress JL, Li N, Wehrens XH (2013) Alterations in ryanodine receptors and related proteins in heart failure. Biochim Biophys Acta 1832:2425–2431. https://doi.org/10.1016/j.bbadis.2013.06.008
Balke CW, Shorofsky SR (1998) Alterations in calcium handling in cardiac hypertrophy and heart failure. Cardiovasc Res 37:290–299. https://doi.org/10.1016/s0008-6363(97)00272-1
Bassani JW, Yuan W, Bers DM (1995) Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am J Physiol 268:C1313–C1319
Beckman KB, Ames BN (1998) The free radical theory of aging matures. Physiol Rev 78:547–581
Belevych A, Kubalova Z, Terentyev D, Hamlin RL, Carnes CA, Györke S (2007) Enhanced ryanodine receptor-mediated calcium leak determines reduced sarcoplasmic reticulum calcium content in chronic canine heart failure. Biophys J 93:4083–4092. https://doi.org/10.1529/biophysj.107.114546
Belmin J, Bernard C, Corman B, Merval R, Esposito B, Tedgui A (1995) Increased production of tumor necrosis factor and interleukin-6 by arterial wall of aged rats. Am J Physiol 268:H2288–H2293
Benjamin EJ, Levy D, Vaziri SM, D’Agostino RB, Belanger AJ, Wolf PA (1994) Independent risk factors for atrial fibrillation in a population-based cohort. The Framingham Heart Study. JAMA 271:840–844
Benkusky NA, Weber CS, Scherman JA, Farrell EF, Hacker TA, John MC, Powers PA, Valdivia HH (2007) Intact beta-adrenergic response and unmodified progression toward heart failure in mice with genetic ablation of a major protein kinase A phosphorylation site in the cardiac ryanodine receptor. Circ Res 101:819–829. https://doi.org/10.1161/circresaha.107.153007
Bers DM (2000) Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res 87:275–281. https://doi.org/10.1161/01.res.87.4.275
Bers DM (2008) Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 70:23–49. https://doi.org/10.1146/annurev.physiol.70.113006.100455
Bers DM (2014) Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu Rev Physiol 76:107–127. https://doi.org/10.1146/annurev-physiol-020911-153308
Blayney LM, Lai FA (2009) Ryanodine receptor-mediated arrhythmias and sudden cardiac death. Pharmacol Ther 123:151–177. https://doi.org/10.1016/j.pharmthera.2009.03.006
Bovo E, Mazurek SR, Zima AV (2018) Oxidation of ryanodine receptor after ischemia-reperfusion increases propensity of Ca(2+) waves during beta-adrenergic receptor stimulation. Am J Physiol Heart Circ Physiol 315:H1032–H1040. https://doi.org/10.1152/ajpheart.00334.2018
Breckenridge R (2010) Heart failure and mouse models. Dis Model Mech 3:138–143. https://doi.org/10.1242/dmm.005017
Cai WF, Liu GS, Lam CK, Florea S, Qian J, Zhao W, Pritchard T, Haghighi K, Lebeche D, Lu LJ, Deng J, Fan GC, Hajjar RJ, Kranias EG (2015) Up-regulation of micro-RNA765 in human failing hearts is associated with post-transcriptional regulation of protein phosphatase inhibitor-1 and depressed contractility. Eur J Heart Fail 17:782–793. https://doi.org/10.1002/ejhf.323
Cardin S, Li D, Thorin-Trescases N, Leung TK, Thorin E, Nattel S (2003) Evolution of the atrial fibrillation substrate in experimental congestive heart failure: angiotensin-dependent and -independent pathways. Cardiovasc Res 60:315–325
Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Müller FU, Schmitz W, Schotten U, Anderson ME, Valderrábano M, Dobrev D, Wehrens XH (2009) Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest 119:1940–1951. https://doi.org/10.1172/jci37059
Chiang DY, Kongchan N, Beavers DL, Alsina KM, Voigt N, Neilson JR, Jakob H, Martin JF, Dobrev D, Wehrens XH, Li N (2014) Loss of microRNA-106b-25 cluster promotes atrial fibrillation by enhancing ryanodine receptor type-2 expression and calcium release. Circ Arrhythm Electrophysiol 7:1214–1222. https://doi.org/10.1161/CIRCEP.114.001973
Chu G, Kranias EG (2006) Phospholamban as a therapeutic modality in heart failure. Novartis Found Symp 274:156–171 discussion 172-155, 272-156
Connell P, Word TA, Wehrens XHT (2020) Targeting pathological leak of ryanodine receptors: preclinical progress and the potential impact on treatments for cardiac arrhythmias and heart failure. Expert Opin Ther Targets 24:25–36. https://doi.org/10.1080/14728222.2020.1708326
Davis RJ (2000) Signal transduction by the JNK group of MAP kinases. Cell 103:239–252. https://doi.org/10.1016/s0092-8674(00)00116-1
Denham NC, Pearman CM, Caldwell JL, Madders GWP, Eisner DA, Trafford AW, Dibb KM (2018) Calcium in the pathophysiology of atrial fibrillation and heart failure. Front Physiol 9:1380. https://doi.org/10.3389/fphys.2018.01380
Desantiago J, Ai X, Islam M, Acuna G, Ziolo MT, Bers DM, Pogwizd SM (2008) Arrhythmogenic effects of beta2-adrenergic stimulation in the failing heart are attributable to enhanced sarcoplasmic reticulum Ca load. Circ Res 102:1389–1397. doi:CIRCRESAHA.107.169011 [pii]. https://doi.org/10.1161/CIRCRESAHA.107.169011
Dhanasekaran DN, Reddy EP (2017) JNK-signaling: a multiplexing hub in programmed cell death. Genes Cancer 8:682–694. https://doi.org/10.18632/genesandcancer.155
Di Salvo TG, Winterfield J (2018) Holiday heart: some sobering mechanistic insights. J Am Coll Cardiol 71:1471–1473. https://doi.org/10.1016/j.jacc.2018.02.009
Dobrev D, Wehrens XHT (2017) Calcium-mediated cellular triggered activity in atrial fibrillation. J Physiol 595:4001–4008. https://doi.org/10.1113/jp273048
Eisner DA, Caldwell JL, Kistamás K, Trafford AW (2017) Calcium and Excitation-Contraction Coupling in the Heart. Circ Res 121:181–195. https://doi.org/10.1161/circresaha.117.310230
El-Armouche A, Boknik P, Eschenhagen T, Carrier L, Knaut M, Ravens U, Dobrev D (2006) Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation 114:670–680. https://doi.org/10.1161/circulationaha.106.636845
El-Mas MM, Fan M, Abdel-Rahman AA (2013) Role of rostral ventrolateral medullary ERK/JNK/p38 MAPK signaling in the pressor effects of ethanol and its oxidative product acetaldehyde. Alcoholism, clinical and experimental research 37:1827–1837. https://doi.org/10.1111/acer.12179
Elamin E, Masclee A, Troost F, Pieters HJ, Keszthelyi D, Aleksa K, Dekker J, Jonkers D (2014) Ethanol impairs intestinal barrier function in humans through mitogen activated protein kinase signaling: a combined in vivo and in vitro approach. PLoS One 9:e107421. https://doi.org/10.1371/journal.pone.0107421
Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O’Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME (2008) A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133:462–474. https://doi.org/10.1016/j.cell.2008.02.048
Erickson JR, Nichols CB, Uchinoumi H, Stein ML, Bossuyt J, Bers DM (2015) S-Nitrosylation Induces Both Autonomous Activation and Inhibition of Calcium/Calmodulin-dependent Protein Kinase II δ. J Biol Chem 290:25646–25656. https://doi.org/10.1074/jbc.M115.650234
Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM (2013) Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 502:372–376. https://doi.org/10.1038/nature12537
Fabiato A (1985) Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol 85:247–289
Feng N, Anderson ME (2017) CaMKII is a nodal signal for multiple programmed cell death pathways in heart. J Mol Cell Cardiol 103:102–109. https://doi.org/10.1016/j.yjmcc.2016.12.007
Fernández-Miranda G, Romero-Garcia T, Barrera-Lechuga TP, Mercado-Morales M, Rueda A (2019) Impaired activity of ryanodine receptors contributes to calcium mishandling in cardiomyocytes of metabolic syndrome rats. Front Physiol 10:520. https://doi.org/10.3389/fphys.2019.00520
Gambardella J, Trimarco B, Iaccarino G, Santulli G (2018) New insights in cardiac calcium handling and excitation-contraction coupling. Adv Exp Med Biol 1067:373–385. https://doi.org/10.1007/5584_2017_106
Gao X, Wu X, Yan J, Zhang J, Zhao W, DeMarco D, Zhang Y, Bakhos M, Mignery G, Sun J, Li Z, Fill M, Ai X (2018) Transcriptional regulation of stress kinase JNK2 in pro-arrhythmic CaMKIIδ expression in the aged atrium. Cardiovasc Res. 114:737–746. https://doi.org/10.1093/cvr/cvy011
Gius D, Botero A, Shah S, Curry HA (1999) Intracellular oxidation/reduction status in the regulation of transcription factors NF-kappaB and AP-1. Toxicol Lett 106:93–106
Glynn P, Musa H, Wu X, Unudurthi SD, Little S, Qian L, Wright PJ, Radwanski PB, Gyorke S, Mohler PJ, Hund TJ (2015) Voltage-gated sodium channel phosphorylation at Ser571 regulates late current, arrhythmia, and cardiac function in vivo. Circulation 132:567–577. https://doi.org/10.1161/circulationaha.114.015218
Go AS, Hylek EM, Phillips KA, Chang Y, Henault LE, Selby JV, Singer DE (2001) Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 285:2370–2375
Gorski PA, Ceholski DK, Hajjar RJ (2015) Altered myocardial calcium cycling and energetics in heart failure--a rational approach for disease treatment. Cell Metab 21:183–194. https://doi.org/10.1016/j.cmet.2015.01.005
Greiser M, Neuberger HR, Harks E, El-Armouche A, Boknik P, de Haan S, Verheyen F, Verheule S, Schmitz W, Ravens U, Nattel S, Allessie MA, Dobrev D, Schotten U (2009) Distinct contractile and molecular differences between two goat models of atrial dysfunction: AV block-induced atrial dilatation and atrial fibrillation. J Mol Cell Cardiol 46:385–394. https://doi.org/10.1016/j.yjmcc.2008.11.012
Gutierrez DA, Fernandez-Tenorio M, Ogrodnik J, Niggli E (2013) NO-dependent CaMKII activation during beta-adrenergic stimulation of cardiac muscle. Cardiovasc Res 100:392–401. https://doi.org/10.1093/cvr/cvt201
Hegyi B, Morotti S, Liu C, Ginsburg KS, Bossuyt J, Belardinelli L, Izu LT, Chen-Izu Y, Bányász T, Grandi E, Bers DM (2019) Enhanced depolarization drive in failing rabbit ventricular myocytes: calcium-dependent and β-adrenergic effects on late sodium, L-type calcium, and sodium-calcium exchange currents. Circ Arrhythm Electrophysiol 12:e007061. https://doi.org/10.1161/circep.118.007061
Heijman J, Dewenter M, El-Armouche A, Dobrev D (2013) Function and regulation of serine/threonine phosphatases in the healthy and diseased heart. J Mol Cell Cardiol 64:90–98. https://doi.org/10.1016/j.yjmcc.2013.09.006
Heijman J, Muna AP, Veleva T, Molina CE, Sutanto H, Tekook M, Wang Q, Abu-Taha IH, Gorka M, Künzel S, El-Armouche A, Reichenspurner H, Kamler M, Nikolaev V, Ravens U, Li N, Nattel S, Wehrens XHT, Dobrev D (2020) Atrial myocyte NLRP3/CaMKII nexus forms a substrate for postoperative atrial fibrillation. Circ Res 127:1036–1055. https://doi.org/10.1161/circresaha.120.316710
Hoch B, Meyer R, Hetzer R, Krause EG, Karczewski P (1999) Identification and expression of delta-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ Res 84:713–721
Houser SR, Piacentino V 3rd, Weisser J (2000) Abnormalities of calcium cycling in the hypertrophied and failing heart. J Mol Cell Cardiol 32:1595–1607. https://doi.org/10.1006/jmcc.2000.1206
Hu MC, Qiu WR, Wang YP (1997) JNK1, JNK2 and JNK3 are p53 N-terminal serine 34 kinases. Oncogene 15:2277–2287. https://doi.org/10.1038/sj.onc.1201401
Hu ST, Shen YF, Liu GS, Lei CH, Tang Y, Wang JF, Yang YJ (2010) Altered intracellular Ca2+ regulation in chronic rat heart failure. J Physiol Sci 60:85–94. https://doi.org/10.1007/s12576-009-0070-6
Huang C, Cao W, Liao R, Wang J, Wang Y, Tong L, Chen X, Zhu W, Zhang W (2014) PP1γ functionally augments the alternative splicing of CaMKIIδ through interaction with ASF. Am J Physiol Cell Physiol 306:C167–C177. https://doi.org/10.1152/ajpcell.00145.2013
Hund TJ, Mohler PJ (2015) Role of CaMKII in cardiac arrhythmias. Trends in cardiovascular medicine 25:392–397. https://doi.org/10.1016/j.tcm.2014.12.001
Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD (2014) Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ Res 114:266–282. https://doi.org/10.1161/CIRCRESAHA.113.301720
Izumi Y, Kim S, Murakami T, Yamanaka S, Iwao H (1998) Cardiac mitogen-activated protein kinase activities are chronically increased in stroke-prone hypertensive rats. Hypertension 31:50–56
Juhaszova M, Rabuel C, Zorov DB, Lakatta EG, Sollott SJ (2005) Protection in the aged heart: preventing the heart-break of old age? Cardiovasc Res 66:233–244. https://doi.org/10.1016/j.cardiores.2004.12.020
Kaoud TS, Yan C, Mitra S, Tseng CC, Jose J, Taliaferro JM, Tuohetahuntila M, Devkota A, Sammons R, Park J, Park H, Shi Y, Hong J, Ren P, Dalby KN (2012) From in silico discovery to intra-cellular activity: targeting JNK-protein interactions with small molecules. ACS Med Chem Lett 3:721–725. https://doi.org/10.1021/ml300129b
Karin M (2005) Inflammation-activated protein kinases as targets for drug development. Proc Am Thorac Soc 2:386–390; discussion 394-385 . https://doi.org/10.1513/pats.200504-034SR2/4/386 [pii]
Karin M, Gallagher E (2005) From JNK to pay dirt: jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life 57:283–295. https://doi.org/10.1080/15216540500097111
Kashimura T, Briston SJ, Trafford AW, Napolitano C, Priori SG, Eisner DA, Venetucci LA (2010) In the RyR2(R4496C) mouse model of CPVT, beta-adrenergic stimulation induces Ca waves by increasing SR Ca content and not by decreasing the threshold for Ca waves. Circ Res 107:1483–1489. https://doi.org/10.1161/CIRCRESAHA.110.227744
Kistamás K, Veress R, Horváth B, Bányász T, Nánási PP, Eisner DA (2020) Calcium handling defects and cardiac arrhythmia syndromes. Front Pharmacol 11:72. https://doi.org/10.3389/fphar.2020.00072
Knight RJ, Buxton DB (1996) Stimulation of c-Jun kinase and mitogen-activated protein kinase by ischemia and reperfusion in the perfused rat heart. Biochem Biophys Res Commun 218:83–88. https://doi.org/10.1006/bbrc.1996.0016
Koval OM, Snyder JS, Wolf RM, Pavlovicz RE, Glynn P, Curran J, Leymaster ND, Dun W, Wright PJ, Cardona N, Qian L, Mitchell CC, Boyden PA, Binkley PF, Li C, Anderson ME, Mohler PJ, Hund TJ (2012) Ca2+/calmodulin-dependent protein kinase II-based regulation of voltage-gated Na+ channel in cardiac disease. Circulation 126:2084–2094. https://doi.org/10.1161/circulationaha.112.105320
Kreusser MM, Lehmann LH, Keranov S, Hoting MO, Oehl U, Kohlhaas M, Reil JC, Neumann K, Schneider MD, Hill JA, Dobrev D, Maack C, Maier LS, Gröne HJ, Katus HA, Olson EN, Backs J (2014) Cardiac CaM Kinase II genes δ and γ contribute to adverse remodeling but redundantly inhibit calcineurin-induced myocardial hypertrophy. Circulation 130:1262–1273. https://doi.org/10.1161/circulationaha.114.006185
Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA (1999) The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron 22:667–676
Kyriakis JM, Avruch J (1990) pp54 microtubule-associated protein 2 kinase. A novel serine/threonine protein kinase regulated by phosphorylation and stimulated by poly-L-lysine. J Biol Chem 265:17355–17363
Kyriakis JM, Brautigan DL, Ingebritsen TS, Avruch J (1991) pp54 microtubule-associated protein-2 kinase requires both tyrosine and serine/threonine phosphorylation for activity. J Biol Chem 266:10043–10046
Landstrom AP, Dobrev D, Wehrens XHT (2017) Calcium signaling and cardiac arrhythmias. Circ Res 120:1969–1993. https://doi.org/10.1161/circresaha.117.310083
Lehnart SE, Schillinger W, Pieske B, Prestle J, Just H, Hasenfuss G (1998) Sarcoplasmic reticulum proteins in heart failure. Ann N Y Acad Sci 853:220–230. https://doi.org/10.1111/j.1749-6632.1998.tb08270.x
Li J, Marionneau C, Zhang R, Shah V, Hell JW, Nerbonne JM, Anderson ME (2006) Calmodulin kinase II inhibition shortens action potential duration by upregulation of K+ currents. Circ Res 99:1092–1099. https://doi.org/10.1161/01.RES.0000249369.71709.5c
Li N, Chiang DY, Wang S, Wang Q, Sun L, Voigt N, Respress JL, Ather S, Skapura DG, Jordan VK, Horrigan FT, Schmitz W, Müller FU, Valderrabano M, Nattel S, Dobrev D, Wehrens XHT (2014) Ryanodine receptor-mediated calcium leak drives progressive development of an atrial fibrillation substrate in a transgenic mouse model. Circulation 129:1276–1285. https://doi.org/10.1161/circulationaha.113.006611
Li SY, Du M, Dolence EK, Fang CX, Mayer GE, Ceylan-Isik AF, LaCour KH, Yang X, Wilbert CJ, Sreejayan N, Ren J (2005) Aging induces cardiac diastolic dysfunction, oxidative stress, accumulation of advanced glycation endproducts and protein modification. Aging Cell 4:57–64. doi:ACE146 [pii]. https://doi.org/10.1111/j.1474-9728.2005.00146.x
Lindner M, Böhle T, Beuckelmann DJ (2002) Ca2+-handling in heart failure--a review focusing on Ca2+ sparks. Basic Res Cardiol 97(Suppl 1):I79–I82. https://doi.org/10.1007/s003950200034
Liu B, Ho HT, Brunello L, Unudurthi SD, Lou Q, Belevych AE, Qian L, Kim do H, Cho C, Janssen PM, Hund TJ, Knollmann BC, Kranias EG, Gyorke S (2015) Ablation of HRC alleviates cardiac arrhythmia and improves abnormal Ca handling in CASQ2 knockout mice prone to CPVT. Cardiovasc Res 108:299–311. https://doi.org/10.1093/cvr/cvv222cvv222 [pii]
Louch WE, Bito V, Heinzel FR, Macianskiene R, Vanhaecke J, Flameng W, Mubagwa K, Sipido KR (2004) Reduced synchrony of Ca2+ release with loss of T-tubules-a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc Res 62:63–73. https://doi.org/10.1016/j.cardiores.2003.12.031
Lugenbiel P, Wenz F, Govorov K, Schweizer PA, Katus HA, Thomas D (2015) Atrial fibrillation complicated by heart failure induces distinct remodeling of calcium cycling proteins. PLoS One 10:e0116395. https://doi.org/10.1371/journal.pone.0116395
MacDonnell SM, García-Rivas G, Scherman JA, Kubo H, Chen X, Valdivia H, Houser SR (2008) Adrenergic regulation of cardiac contractility does not involve phosphorylation of the cardiac ryanodine receptor at serine 2808. Circ Res 102:e65–e72. https://doi.org/10.1161/circresaha.108.174722
Maier LS (2005) CaMKIIdelta overexpression in hypertrophy and heart failure: cellular consequences for excitation-contraction coupling. Braz J Med Biol Res 38:1293–1302. https://doi.org/10.1590/s0100-879x2005000900002
Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH, Bers DM (2003) Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res 92:904–911. https://doi.org/10.1161/01.RES.0000069685.20258.F1
Makarewich CA, Munir AZ, Schiattarella GG, Bezprozvannaya S, Raguimova ON, Cho EE, Vidal AH, Robia SL, Bassel-Duby R, Olson EN (2018) The DWORF micropeptide enhances contractility and prevents heart failure in a mouse model of dilated cardiomyopathy. Elife 7:7. https://doi.org/10.7554/eLife.38319
Marks AR (2013) Calcium cycling proteins and heart failure: mechanisms and therapeutics. J Clin Invest 123:46–52. https://doi.org/10.1172/jci62834
Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR (2000) PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101:365–376. https://doi.org/10.1016/s0092-8674(00)80847-8
Maxwell JT, Natesan S, Mignery GA (2012) Modulation of inositol 1,4,5-trisphosphate receptor type 2 channel activity by Ca2+/calmodulin-dependent protein kinase II (CaMKII)-mediated phosphorylation. J Biol Chem 287:39419–39428. https://doi.org/10.1074/jbc.M112.374058
Meyer-Roxlau S, Lämmle S, Opitz A, Künzel S, Joos JP, Neef S, Sekeres K, Sossalla S, Schöndube F, Alexiou K, Maier LS, Dobrev D, Guan K, Weber S, El-Armouche A (2017) Differential regulation of protein phosphatase 1 (PP1) isoforms in human heart failure and atrial fibrillation. Basic Res Cardiol 112:43. https://doi.org/10.1007/s00395-017-0635-0
Minamisawa S, Sato Y, Cho MC (2004) Calcium cycling proteins in heart failure, cardiomyopathy and arrhythmias. Exp Mol Med 36:193–203. https://doi.org/10.1038/emm.2004.27
Mohammad J, Singh RR, Riggle C, Haugrud B, Abdalla MY, Reindl KM (2019) JNK inhibition blocks piperlongumine-induced cell death and transcriptional activation of heme oxygenase-1 in pancreatic cancer cells. Apoptosis 24:730–744. https://doi.org/10.1007/s10495-019-01553-9
Mohit AA, Martin JH, Miller CA (1995) p493F12 kinase: a novel MAP kinase expressed in a subset of neurons in the human nervous system. Neuron 14:67–78. https://doi.org/10.1016/0896-6273(95)90241-4
Molina CE, Abu-Taha IH, Wang Q, Roselló-Díez E, Kamler M, Nattel S, Ravens U, Wehrens XHT, Hove-Madsen L, Heijman J, Dobrev D (2018) Profibrotic, electrical, and calcium-handling remodeling of the atria in heart failure patients with and without atrial fibrillation. Front Physiol 9:1383. https://doi.org/10.3389/fphys.2018.01383
Morooka H, Bonventre JV, Pombo CM, Kyriakis JM, Force T (1995) Ischemia and reperfusion enhance ATF-2 and c-Jun binding to cAMP response elements and to an AP-1 binding site from the c-jun promoter. J Biol Chem 270:30084–30092. https://doi.org/10.1074/jbc.270.50.30084
Mustroph J, Maier LS, Wagner S (2014) CaMKII regulation of cardiac K channels. Front Pharmacol 5:20. https://doi.org/10.3389/fphar.2014.00020
Nakano R, Nakayama T, Sugiya H (2020) Biological Properties of JNK3 and Its Function in Neurons, Astrocytes, Pancreatic β-Cells and Cardiovascular Cells. Cells 9. https://doi.org/10.3390/cells9081802
Neef S, Dybkova N, Sossalla S, Ort KR, Fluschnik N, Neumann K, Seipelt R, Schondube FA, Hasenfuss G, Maier LS (2010) CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ Res 106:1134–1144. doi:CIRCRESAHA.109.203836 [pii]. https://doi.org/10.1161/CIRCRESAHA.109.203836
Neef S, Heijman J, Otte K, Dewenter M, Saadatmand AR, Meyer-Roxlau S, Antos CL, Backs J, Dobrev D, Wagner M, Maier LS, El-Armouche A (2017) Chronic loss of inhibitor-1 diminishes cardiac RyR2 phosphorylation despite exaggerated CaMKII activity. Naunyn Schmiedebergs Arch Pharmacol 390:857–862. https://doi.org/10.1007/s00210-017-1376-1
Nerbonne JM (2011) Repolarizing cardiac potassium channels: multiple sites and mechanisms for CaMKII-mediated regulation. Heart Rhythm 8:938–941. https://doi.org/10.1016/j.hrthm.2011.01.018
Neuman RB, Bloom HL, Shukrullah I, Darrow LA, Kleinbaum D, Jones DP, Dudley SC Jr (2007) Oxidative stress markers are associated with persistent atrial fibrillation. Clin Chem 53:1652–1657. doi:clinchem.2006.083923 [pii]. https://doi.org/10.1373/clinchem.2006.083923
Nishitani Y, Matsumoto H (2006) Ethanol rapidly causes activation of JNK associated with ER stress under inhibition of ADH. FEBS letters 580:9–14. https://doi.org/10.1016/j.febslet.2005.11.030
Ojeda ML, Barrero MJ, Nogales F, Murillo ML, Carreras O (2012) Oxidative effects of chronic ethanol consumption on the functions of heart and kidney: folic acid supplementation. Alcohol Alcohol 47:404–412. https://doi.org/10.1093/alcalc/ags056
Omura T, Yoshiyama M, Shimada T, Shimizu N, Kim S, Iwao H, Takeuchi K, Yoshikawa J (1999) Activation of mitogen-activated protein kinases in in vivo ischemia/reperfused myocardium in rats. J Mol Cell Cardiol 31:1269–1279. https://doi.org/10.1006/jmcc.1999.0959
Ottolia M, Torres N, Bridge JH, Philipson KD, Goldhaber JI (2013) Na/Ca exchange and contraction of the heart. J Mol Cell Cardiol 61:28–33. https://doi.org/10.1016/j.yjmcc.2013.06.001
Peart JN, Gross ER, Headrick JP, Gross GJ (2007) Impaired p38 MAPK/HSP27 signaling underlies aging-related failure in opioid-mediated cardioprotection. J Mol Cell Cardiol 42:972–980. doi:S0022-2828(07)00050-8 [pii]. https://doi.org/10.1016/j.yjmcc.2007.02.011
Pombo CM, Bonventre JV, Avruch J, Woodgett JR, Kyriakis JM, Force T (1994) The stress-activated protein kinases are major c-Jun amino-terminal kinases activated by ischemia and reperfusion. J Biol Chem 269:26546–26551
Reiken S, Lacampagne A, Zhou H, Kherani A, Lehnart SE, Ward C, Huang F, Gaburjakova M, Gaburjakova J, Rosemblit N, Warren MS, He KL, Yi GH, Wang J, Burkhoff D, Vassort G, Marks AR (2003) PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: defective regulation in heart failure. J Cell Biol 160:919–928. https://doi.org/10.1083/jcb.200211012
Respress JL, van Oort RJ, Li N, Rolim N, Dixit SS, deAlmeida A, Voigt N, Lawrence WS, Skapura DG, Skårdal K, Wisløff U, Wieland T, Ai X, Pogwizd SM, Dobrev D, Wehrens XH (2012) Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ Res 110:1474–1483. https://doi.org/10.1161/circresaha.112.268094
Rich MW (2009) Epidemiology of atrial fibrillation. J Interv Card Electrophysiol 25:3–8. https://doi.org/10.1007/s10840-008-9337-8
Rokita AG, Anderson ME (2012) New therapeutic targets in cardiology: arrhythmias and Ca2+/calmodulin-dependent kinase II (CaMKII). Circulation 126:2125–2139. https://doi.org/10.1161/circulationaha.112.124990
Ronkainen JJ, Hänninen SL, Korhonen T, Koivumäki JT, Skoumal R, Rautio S, Ronkainen VP, Tavi P (2011) Ca2+-calmodulin-dependent protein kinase II represses cardiac transcription of the L-type calcium channel alpha(1C)-subunit gene (Cacna1c) by DREAM translocation. J Physiol 589:2669–2686. https://doi.org/10.1113/jphysiol.2010.201400
Rose BA, Force T, Wang Y (2010) Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev 90:1507–1546. https://doi.org/10.1152/physrev.00054.2009
Rougier O, Vassort G, Garnier D, Gargouil YM, Coraboeuf E (1969) Existence and role of a slow inward current during the frog atrial action potential. Pflugers Arch 308:91–110
Santulli G, Nakashima R, Yuan Q, Marks AR (2017) Intracellular calcium release channels: an update. J Physiol 595:3041–3051. https://doi.org/10.1113/jp272781
Santulli G, Xie W, Reiken SR, Marks AR (2015) Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci U S A 112:11389–11394. https://doi.org/10.1073/pnas.1513047112
Schaeffer HJ, Weber MJ (1999) Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol 19:2435–2444. https://doi.org/10.1128/mcb.19.4.2435
Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Meli AC, Wronska A, Dura M, Chen BX, Marks AR (2010) Role of chronic ryanodine receptor phosphorylation in heart failure and β-adrenergic receptor blockade in mice. J Clin Invest 120:4375–4387. https://doi.org/10.1172/jci37649
Shannon TR, Ginsburg KS, Bers DM (2000) Potentiation of fractional sarcoplasmic reticulum calcium release by total and free intra-sarcoplasmic reticulum calcium concentration. Biophys J 78:334–343. https://doi.org/10.1016/S0006-3495(00)76596-9
Shannon TR, Pogwizd SM, Bers DM (2003) Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ Res 93:592–594. https://doi.org/10.1161/01.res.0000093399.11734.b3
Shiferaw Y, Watanabe MA, Garfinkel A, Weiss JN, Karma A (2003) Model of intracellular calcium cycling in ventricular myocytes. Biophys J 85:3666–3686. https://doi.org/10.1016/s0006-3495(03)74784-5
Simmerman HK, Jones LR (1998) Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physiol Rev 78:921–947. https://doi.org/10.1152/physrev.1998.78.4.921
Sjaastad I, Wasserstrom JA, Sejersted OM (2003) Heart failure -- a challenge to our current concepts of excitation-contraction coupling. J Physiol 546:33–47. https://doi.org/10.1113/jphysiol.2002.034728
Smith GL, Eisner DA (2019) Calcium buffering in the heart in health and disease. Circulation 139:2358–2371. https://doi.org/10.1161/circulationaha.118.039329
Sossalla S, Fluschnik N, Schotola H, Ort KR, Neef S, Schulte T, Wittkopper K, Renner A, Schmitto JD, Gummert J, El-Armouche A, Hasenfuss G, Maier LS (2010) Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ Res 107:1150–1161. https://doi.org/10.1161/CIRCRESAHA.110.220418
Sugden PH, Clerk A (1998) “Stress-Responsive” Mitogen-Activated Protein Kinases (c-Jun N-Terminal Kinases and p38 Mitogen-Activated Protein Kinases) in the Myocardium. Circ Res 83:345–352. https://doi.org/10.1161/01.res.83.4.345
Sun L, Chen Y, Luo H, Xu M, Meng G, Zhang W (2019) Ca(2+)/calmodulin-dependent protein kinase II regulation by inhibitor 1 of protein phosphatase 1 alleviates necroptosis in high glucose-induced cardiomyocytes injury. Biochem Pharmacol 163:194–205. https://doi.org/10.1016/j.bcp.2019.02.022
Tamayo M, Fulgencio-Covián A, Navarro-García JA, Val-Blasco A, Ruiz-Hurtado G, Gil-Fernández M, Martín-Nunes L, Lopez JA, Desviat LR, Delgado C, Richard E, Fernández-Velasco M (1866) Intracellular calcium mishandling leads to cardiac dysfunction and ventricular arrhythmias in a mouse model of propionic acidemia. Biochim Biophys Acta Mol Basis Dis 2020:165586. https://doi.org/10.1016/j.bbadis.2019.165586
Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu-Taha I, Sun Q, Wieland T, Ravens U, Nattel S, Wehrens XH, Dobrev D (2012) Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 125:2059–2070. https://doi.org/10.1161/circulationaha.111.067306
Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS (2006) Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest 116:3127–3138. https://doi.org/10.1172/jci26620
Wagner S, Hacker E, Grandi E, Weber SL, Dybkova N, Sossalla S, Sowa T, Fabritz L, Kirchhof P, Bers DM, Maier LS (2009) Ca/calmodulin kinase II differentially modulates potassium currents. Circ Arrhythm Electrophysiol 2:285–294. https://doi.org/10.1161/circep.108.842799
Wakili R, Yeh YH, Yan Qi X, Greiser M, Chartier D, Nishida K, Maguy A, Villeneuve LR, Boknik P, Voigt N, Krysiak J, Kääb S, Ravens U, Linke WA, Stienen GJ, Shi Y, Tardif JC, Schotten U, Dobrev D, Nattel S (2010) Multiple potential molecular contributors to atrial hypocontractility caused by atrial tachycardia remodeling in dogs. Circ Arrhythm Electrophysiol 3:530–541. https://doi.org/10.1161/circep.109.933036
Walweel K, Molenaar P, Imtiaz MS, Denniss A, Dos Remedios C, van Helden DF, Dulhunty AF, Laver DR, Beard NA (2017) Ryanodine receptor modification and regulation by intracellular Ca(2+) and Mg(2+) in healthy and failing human hearts. J Mol Cell Cardiol 104:53–62. https://doi.org/10.1016/j.yjmcc.2017.01.016
Wang MC, Bohmann D, Jasper H (2003) JNK signaling confers tolerance to oxidative stress and extends lifespan in Drosophila. Dev Cell 5:811–816
Wang Y, Tandan S, Cheng J, Yang C, Nguyen L, Sugianto J, Johnstone JL, Sun Y, Hill JA (2008) Ca2+/calmodulin-dependent protein kinase II-dependent remodeling of Ca2+ current in pressure overload heart failure. J Biol Chem 283:25524–25532. https://doi.org/10.1074/jbc.M803043200
Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR (2006) Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci U S A 103:511–518. https://doi.org/10.1073/pnas.0510113103
Wescott AP, Jafri MS, Lederer WJ, Williams GS (2016) Ryanodine receptor sensitivity governs the stability and synchrony of local calcium release during cardiac excitation-contraction coupling. J Mol Cell Cardiol 92:82–92. https://doi.org/10.1016/j.yjmcc.2016.01.024
Xiao B, Sutherland C, Walsh MP, Chen SR (2004) Protein kinase A phosphorylation at serine-2808 of the cardiac Ca2+-release channel (ryanodine receptor) does not dissociate 12.6-kDa FK506-binding protein (FKBP12.6). Circ Res 94:487–495. https://doi.org/10.1161/01.res.0000115945.89741.22
Xiao L, Coutu P, Villeneuve LR, Tadevosyan A, Maguy A, Le Bouter S, Allen BG, Nattel S (2008) Mechanisms underlying rate-dependent remodeling of transient outward potassium current in canine ventricular myocytes. Circ Res 103:733–742. https://doi.org/10.1161/circresaha.108.171157
Xie LH, Sato D, Garfinkel A, Qu Z, Weiss JN (2008) Intracellular Ca alternans: coordinated regulation by sarcoplasmic reticulum release, uptake, and leak. Biophys J 95:3100–3110. https://doi.org/10.1529/biophysj.108.130955
Xu L, Chen J, Li XY, Ren S, Huang CX, Wu G, Jiang XJ (2012) Analysis of Na(+)/Ca (2+) exchanger (NCX) function and current in murine cardiac myocytes during heart failure. Mol Biol Rep 39:3847–3852. https://doi.org/10.1007/s11033-011-1163-x
Yan J, Kong W, Zhang Q, Beyer EC, Walcott G, Fast VG, Ai X (2013) c-Jun N-terminal kinase activation contributes to reduced connexin43 and development of atrial arrhythmias. Cardiovasc Res 97:589–597. https://doi.org/10.1093/cvr/cvs366
Yan J, Thomson JK, Zhao W, Gao X, Huang F, Chen B, Liang Q, Song LS, Fill M, Ai X (2018) Role of stress kinase JNK in binge alcohol-evoked atrial arrhythmia. J Am Coll Cardiol 71:1459–1470. https://doi.org/10.1016/j.jacc.2018.01.060
Yan J, Thomson JK, Zhao W, Wu X, Gao X, DeMarco D, Kong W, Tong M, Sun J, Bakhos M, Fast VG, Liang Q, Prabhu SD, Ai X (2017) The stress kinase JNK regulates gap junction Cx43 gene expression and promotes atrial fibrillation in the aged heart. J Mol Cell Cardiol 114:105–115. https://doi.org/10.1016/j.yjmcc.2017.11.006
Yan J, Zhao W, Thomson JK, Gao X, DeMarco DM, Carrillo E, Chen B, Wu X, Ginsburg KS, Bakhos M, Bers DM, Anderson ME, Song LS, Fill M, Ai X (2018) Stress signaling JNK2 crosstalk with CaMKII underlies enhanced atrial arrhythmogenesis. Circ Res 122:821–835. https://doi.org/10.1161/circresaha.117.312536
Yang L, Wu D, Wang X, Cederbaum AI (2012) Cytochrome P4502E1, oxidative stress, JNK, and autophagy in acute alcohol-induced fatty liver. Free Radical Biology and Medicine 53:1170–1180. https://doi.org/10.1016/j.freeradbiomed.2012.06.029
Yang Y, He J, Yuan M, Tse G, Zhang K, Ma Z, Li J, Zhang Y, Gao Y, Wang R, Li G, Liu T (2020) Xanthine oxidase inhibitor allopurinol improves atrial electrical remodeling in diabetic rats by inhibiting CaMKII/NCX signaling. Life sciences 259:118290. https://doi.org/10.1016/j.lfs.2020.118290
Yeh YH, Wakili R, Qi XY, Chartier D, Boknik P, Kääb S, Ravens U, Coutu P, Dobrev D, Nattel S (2008) Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ Arrhythm Electrophysiol 1:93–102. https://doi.org/10.1161/circep.107.754788
Yoon JY, Ho WK, Kim ST, Cho H (2009) Constitutive CaMKII activity regulates Na+ channel in rat ventricular myocytes. J Mol Cell Cardiol 47:475–484. https://doi.org/10.1016/j.yjmcc.2009.06.020
Yu J, Chen Y, Xu M, Sun L, Luo H, Bao X, Meng G, Zhang W (2019) Ca2+/Calmodulin-Dependent Protein Kinase II Regulation by Inhibitor 1 of Protein Phosphatase 1 Protects Against Myocardial Ischemia–Reperfusion Injury. J Cardiovasc Pharmacol Ther 24:460–473. https://doi.org/10.1177/1074248419841626
Zeke A, Misheva M, Reményi A, Bogoyevitch MA (2016) JNK signaling: regulation and functions based on complex protein-protein partnerships. Microbiol Mol Biol Rev 80:793–835. https://doi.org/10.1128/mmbr.00043-14
Zhang H, Makarewich CA, Kubo H, Wang W, Duran JM, Li Y, Berretta RM, Koch WJ, Chen X, Gao E, Valdivia HH, Houser SR (2012) Hyperphosphorylation of the cardiac ryanodine receptor at serine 2808 is not involved in cardiac dysfunction after myocardial infarction. Circ Res 110:831–840. https://doi.org/10.1161/circresaha.111.255158
Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE Jr, Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME (2005) Calmodulin kinase II inhibition protects against structural heart disease. Nat Med 11:409–417. https://doi.org/10.1038/nm1215
Zhang T, Guo T, Mishra S, Dalton ND, Kranias EG, Peterson KL, Bers DM, Brown JH (2010) Phospholamban ablation rescues sarcoplasmic reticulum Ca(2+) handling but exacerbates cardiac dysfunction in CaMKIIdelta(C) transgenic mice. Circ Res 106:354–362. https://doi.org/10.1161/CIRCRESAHA.109.207423CIRCRESAHA.109.207423 [pii]
Zhang T, Johnson EN, Gu Y, Morissette MR, Sah VP, Gigena MS, Belke DD, Dillmann WH, Rogers TB, Schulman H, Ross J Jr, Brown JH (2002) The cardiac-specific nuclear delta(B) isoform of Ca2+/calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. J Biol Chem 277:1261–1267. https://doi.org/10.1074/jbc.M108525200
Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J Jr, Bers DM, Brown JH (2003) The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res 92:912–919. https://doi.org/10.1161/01.RES.0000069686.31472.C5
Zhang W, Zhang Y, Zhang H, Zhao Q, Liu Z, Xu Y (2019) USP49 inhibits ischemia-reperfusion-induced cell viability suppression and apoptosis in human AC16 cardiomyocytes through DUSP1-JNK1/2 signaling. J Cell Physiol 234:6529–6538. https://doi.org/10.1002/jcp.27390
Zima AV, Bovo E, Mazurek SR, Rochira JA, Li W, Terentyev D (2014) Ca handling during excitation-contraction coupling in heart failure. Pflugers Arch 466:1129–1137. https://doi.org/10.1007/s00424-014-1469-3
Code availability
Not applicable.
Funding
This work was supported by National Institutes of Health (P01-HL06426, R01-AA024769, R01-HL113640, R01-HL146744, and AA024769S2 to XA).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue on Calcium Signal Dynamics in Cardiac Myocytes and Fibroblasts: Mechanisms in Pflügers Archiv—European Journal of Physiology
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
McKee, C., Bare, D.J. & Ai, X. Stress-driven cardiac calcium mishandling via a kinase-to-kinase crosstalk. Pflugers Arch - Eur J Physiol 473, 363–375 (2021). https://doi.org/10.1007/s00424-021-02533-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-021-02533-2