
Abstract
Introduction
Idiopathic pulmonary fibrosis (IPF) is considered a disease of older patients, being rare in patients ≤ 50 years. Still, IPF can occur in younger patients, but this particular patient group is not well characterised so far. The aim of this study was to compare the diagnostic certainty, clinical features, comorbidities and survival in young versus older IPF patients.
Methods
We reviewed our medical records from February 2011 until February 2015, to identify IPF patients, who were then classified as young (≤ 50 years) or older IPF (> 50 years). Radiographic and histological findings, lung function parameters, comorbidities, disease progression and survival were analysed and compared between the two groups.
Results
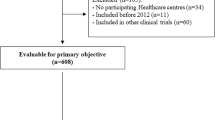
Of 440 patients with interstitial lung disease, 129 patients with IPF were identified, including 30 (23.3%) ≤50 years and 99 (76.7%) > 50 years. There were no differences between age groups in baseline demographics; younger patients were less likely to have a confirmed diagnosis by high-resolution computed tomography (p = 0.014), more likely to require a biopsy (p = 0.08) and less likely to have received antifibrotic therapy (p = 0.006). Despite an overall limited prognosis, younger patients had a significantly better median survival after diagnosis (p = 0.0375), with a significantly higher proportion of older patients dying due to respiratory failure (p = 0.0383).
Conclusion
IPF patients under the age of 50 years have similar features and clinical course compared to older IPF patients. These patients should be diagnosed by adopting a multidisciplinary team approach, potentially benefitting from earlier intervention with effective antifibrotic therapy.




Similar content being viewed by others


Abbreviations
- CTD:
-
Connective tissue disease
- CTD-ILD:
-
Connective tissue disease associated interstitial lung disease
- DLCO:
-
Diffusing capacity for carbon monoxide
- FEV1:
-
Forced expiratory volume in 1 s
- FVC:
-
Forced vital capacity
- GAP:
-
Gender-age-physiology index
- HRCT:
-
High-resolution computed tomography
- ILD:
-
Interstitial lung disease
- IPF:
-
Idiopathic pulmonary fibrosis
- TLC:
-
Total lung capacity
- UIP:
-
Usual interstitial pneumonia
- VC:
-
Vital capacity
- 6MWD:
-
6-min walking distance
References
Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA et al (2011) An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183:788–824
Nathan SD, Shlobin OA, Weir N, Ahmad S, Kaldjob JM, Battle E, Sheridan MJ, du Bois RM (2011) Long-term course and prognosis of idiopathic pulmonary fibrosis in the new millennium. Chest 140:221–229
Gribbin J, Hubbard RB, Le Jeune I, Smith CJ, West J, Tata LJ (2006) Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax 61:980–985
Behr J, Gunther A, Ammenwerth W, Bittmann I, Bonnet R, Buhl R, Eickelberg O, Ewert R, Glaser S, Gottlieb J et al (2013) [German guideline for diagnosis and management of idiopathic pulmonary fibrosis]. Pneumologie 67:81–111
Nadrous HF, Myers JL, Decker PA, Ryu JH (2005) Idiopathic pulmonary fibrosis in patients younger than 50 years. Mayo Clin Proc 80:37–40
Neurohr C, Behr J (2015) Changes in the current classification of IIP: a critical review. Respirology 20:699–704
Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, Lee JS, Leslie KO, Lynch DA, Matteson EL et al (2015) An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J 46:976–987
McEvoy JD, Jones NL (1975) Arterialized capillary blood gases in exercise studies. Med Sci Sports 7:312–315
Laboratories, ATSCoPSfCPF (2002) ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med 166:111–117
Ley B, Ryerson CJ, Vittinghoff E, Ryu JH, Tomassetti S, Lee JS, Poletti V, Buccioli M, Elicker BM, Jones KD et al (2012) A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 156:684–691
Hoeper MM, Bogaard HJ, Condliffe R, Frantz R, Khanna D, Kurzyna M, Langleben D, Manes A, Satoh T, Torres F et al (2013) Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol 62:D42-50
Leuchte HH, Holzapfel M, Baumgartner RA, Ding I, Neurohr C, Vogeser M, Kolbe T, Schwaiblmair M, Behr J (2004) Clinical significance of brain natriuretic peptide in primary pulmonary hypertension. J Am Coll Cardiol 43:764–770
Zimmermann GS, von Wulffen W, Huppmann P, Meis T, Ihle F, Geiseler J, Leuchte HH, Tufman A, Behr J, Neurohr C (2014) Haemodynamic changes in pulmonary hypertension in patients with interstitial lung disease treated with PDE-5 inhibitors. Respirology 19:700–706
Moua T, Maldonado F, Decker PA, Daniels CE, Ryu JH (2014) Frequency and implication of autoimmune serologies in idiopathic pulmonary fibrosis. Mayo Clin Proc 89:319–326
Kreuter M, Ehlers-Tenenbaum S, Palmowski K, Bruhwyler J, Oltmanns U, Muley T, Heussel CP, Warth A, Kolb M, Herth FJ (2016) Impact of Comorbidities on Mortality in Patients with Idiopathic Pulmonary Fibrosis. PLoS ONE 11:e0151425
Raghu G, Amatto VC, Behr J, Stowasser S (2015) Comorbidities in idiopathic pulmonary fibrosis patients: a systematic literature review. Eur Respir J 46:1113–1130
King TE Jr, Tooze JA, Schwarz MI, Brown KR, Cherniack RM (2001) Predicting survival in idiopathic pulmonary fibrosis: scoring system and survival model. Am J Respir Crit Care Med 164:1171–1181
Wells AU, Desai SR, Rubens MB, Goh NS, Cramer D, Nicholson AG, Colby TV, du Bois RM, Hansell DM (2003) Idiopathic pulmonary fibrosis: a composite physiologic index derived from disease extent observed by computed tomography. Am J Respir Crit Care Med 167:962–969
Wang P, Jones KD, Urisman A, Elicker BM, Urbania T, Johannson KA, Assayag D, Lee J, Wolters PJ, Collard HR, Koth LL (2017) Pathologic findings and prognosis in a large prospective cohort of chronic hypersensitivity pneumonitis. Chest 152(3):502–509
Oldham JM, Adegunsoye A, Valenzi E, Lee C, Witt L, Chen L, Husain AN, Montner S, Chung JH, Cottin V et al (2016) Characterisation of patients with interstitial pneumonia with autoimmune features. Eur Respir J 47:1767–1775
Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G (2006) Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 174:810–816
Harari S, Madotto F, Caminati A, Conti S, Cesana G (2016) Epidemiology of idiopathic pulmonary fibrosis in Northern Italy. PLoS ONE 11:e0147072
Acknowledgements
David Young, a professional medical writer from Young Medical Communications and Consulting Limited, critically language-edited the manuscript prior to submission. This support was funded by Ludwig-Maximilian University Munich. We further thank the INSIGHTS-IPF registry for providing comparative data on the prevalence of young IPF patients registered in Germany.
Funding
Ludwig-Maximilian University Munich funded professional writing for this study but other than that there was no funding source involved in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest connected with this work.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed Consent
For this type of study formal consent is not required.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Leuschner, G., Reiter, F., Stocker, F. et al. Idiopathic Pulmonary Fibrosis Among Young Patients: Challenges in Diagnosis and Management. Lung 196, 401–408 (2018). https://doi.org/10.1007/s00408-018-0123-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00408-018-0123-9