
Abstract
Ulotaront is a trace amine-associated receptor 1 (TAAR1) agonist in Phase 3 clinical development for the treatment of schizophrenia. Ulotaront was discovered through a unique, target-agnostic approach optimized to identify drug candidates lacking D2 and 5-HT2A receptor antagonism, while demonstrating an antipsychotic-like phenotypic profile in vivo. The mechanism of action (MOA) of ulotaront is thought to be mediated by agonism at TAAR1 and serotonin 5-HT1A receptors. Ulotaront has completed two Phase 2 trials (4-week acute study and 26-week open-label extension) which led to Breakthrough Therapy Designation from the US Food and Drug Administration for the treatment of schizophrenia. In the double-blind, placebo-controlled, acute study, ulotaront was associated with significant (p < 0.001) improvement in Positive and Negative Syndrome Scale (PANSS) total score (effect size [ES]: 0.45), with improvements vs. placebo also observed across secondary endpoints. Post-hoc analyses of the acute trial revealed additional evidence to support the effect of ulotaront on negative symptoms. In the 4-week study, ulotaront was well-tolerated, with an incidence of adverse events (AEs) numerically lower compared to placebo (45.8% vs. 50.4%; with a number needed to harm [NNH] for individual ulotaront AEs all > 40). The open-label extension demonstrated further improvement across schizophrenia symptoms and confirmed the tolerability of ulotaront, with a 6-month completion rate of 67%. Based on current data, ulotaront shows potential to be a first-in-class TAAR1 agonist for the treatment of schizophrenia with a safety and efficacy profile distinct from current antipsychotics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction: unmet needs in schizophrenia
Schizophrenia is a chronic, multifaceted disorder that consists of symptoms largely grouped into positive, negative, and cognitive domains [1]. Onset typically occurs in late adolescence or early adulthood, and for most patients is characterized by a high degree of chronicity characterized by multiple relapses and remissions (typically partial). [2,3,4]. While lifetime prevalence of schizophrenia is approximately 1%, the early onset, illness chronicity, and degree of associated functional impairment result in the disorder being ranked among the leading causes of disability and economic burden worldwide [5, 6].
For the past 70 years, the treatment of schizophrenia has relied on antipsychotic drugs whose primary mechanism of action (MOA) is via blockade of the dopamine, type 2 (D2) receptor [7, 8]. Second-generation (atypical) antipsychotics (SGAs), introduced almost 30 years after the first-generation antipsychotics (FGAs), also act primarily via a dopamine inhibiting MOA; however, SGAs are also characterized by antagonist activity at the serotonin 5-HT2A receptor [9]. Except for clozapine, SGAs are not associated with significant improvement in efficacy compared to first-generation antipsychotics [10]. Treatment with high potency SGAs and FGAs can be associated with an increased prevalence of movement disorder symptoms and hyperprolactinemia. Additionally, weight gain and adverse metabolic effects with many SGAs and low potency FGAs may contribute to the increased cardiovascular morbidity and mortality [11, 12].
Among individuals with schizophrenia, it is estimated that approximately one-third are non-responders to currently available antipsychotics, while the majority achieve only partial symptom control [13]. In addition to failing to control positive symptoms in some treatment-resistant patients, relapse rates remain high among patients with schizophrenia taking antipsychotics [1]. Due to the high risk of relapse in this patient population, treatment guidelines consistently recommend maintenance therapy with antipsychotics [14]. Unfortunately, for a large proportion of patients, the benefit-risk profile is unfavorable for both first- and second-generation antipsychotics, resulting in discontinuation rates at one year of greater than 50% [15], though use of long-acting injectable antipsychotics may improve long-term adherence [16]. Each relapse makes remission more difficult to achieve, and failure to prevent relapse leads to consequences in both patient health and disease trajectory.
Negative and cognitive symptoms are the least responsive to antipsychotic treatment, while contributing, in large measure, to the comorbidity, poor health-related quality of life, and chronic disability associated with schizophrenia [1, 17,18,19,20,21,22,23]. These two symptom domains are clinically present in the majority of patients with schizophrenia. In an analysis of 20 randomized clinical trials in schizophrenia [24], 62% of patients presented with “prominent” negative symptoms (multiple negative symptoms that were moderate-or-greater in severity); in 20–40% of patients, negative symptoms become a persistent feature of their illness [25]. Clinically significant cognitive impairment exhibits a prevalence in schizophrenia that is comparable to, and as persistent as, negative symptoms, occurring in more than 50% of patients [26, 27]. Reasons for the lack of efficacy of first- and second-generation antipsychotics in effectively treating negative symptoms and cognitive impairment in schizophrenia is uncertain. It has been hypothesized that reduced dopaminergic function in the frontal cortex (and possibly alterations in serotonergic and glutamatergic neurotransmission) may be the neural substrates underlying these two symptom domains [28]. Thus, currently available antipsychotics, acting via antagonism or partial agonism at the D2 receptor, would not be expected to enhance dopaminergic function in the frontal cortex, and in fact, negative symptoms specifically are often observed to be a medication side effect [29].
A substantial investment of research and development resources has been made over the past decade in an attempt to develop drugs with novel, non-D2 MOAs for the treatment of schizophrenia, with particular focus placed on negative and cognitive symptom domains, in addition to an improved tolerability and safety profile [30]. To date, these research efforts have met with little success. However, adjunctive treatments that target specific domains, such as the GlyT1 inhibitor iclepertin, have shown potential [31].
The aim of the current review article is to present an overview of the preclinical and clinical data to date for ulotaront, a novel trace amine-associated receptor 1 (TAAR1) agonist in development for the treatment of schizophrenia, with potential to be one of the first drugs with a non-D2 MOA.
Discovery and characterization of ulotaront
Ulotaront is a trace amine-associated receptor 1 (TAAR1) agonist with additional agonism at 5-HT1A receptors currently in Phase III clinical development for the treatment of schizophrenia. Ulotaront recently reached recommended status for its proposed International Nonproprietary Name (INN), joining TAAR1 partial agonist ralmitaront in the “-taront” class of medications. Ulotaront is the first agent in this novel class of compounds to demonstrate clinical efficacy in a randomized, double-blind, placebo-controlled Phase 2 trial in patients with acute schizophrenia, thus representing a potential “new treatment paradigm” [1]. Based on Phase 2 data, ulotaront was granted Breakthrough Therapy Designation from the U.S. Food and Drug Administration for the treatment of schizophrenia.
Ulotaront was discovered through a unique, target-agnostic approach designed to identify drug candidates that lack D2 and 5-HT2A receptor antagonism yet retain an antipsychotic-like behavioral profile when evaluated in rodents in vivo [32]. In brief, through iterations of the general screening process outlined in Fig. 1A followed by secondary assays, ulotaront was selected for further development [32]. In the in vivo phenotypic screening platform, SmartCube®, ulotaront demonstrated a predominantly antipsychotic-like behavioral profile, with some secondary anxiolytic-, and antidepressant-like activity (Fig. 1B) [32]. Ulotaront is the first, and currently only, compound discovered through this target-agnostic approach to advance to proof-of-concept clinical studies [33, 34].

Discovery and characterization of ulotaront. A Ulotaront discovery. Ulotaront was discovered through a unique, target-agnostic approach designed to identify drug candidates that lack D2 and 5-HT2A receptor antagonism but retain an antipsychotic-like behavioral profile in vivo. B Ulotaront mouse SmartCube® profile. Ulotaront demonstrated a predominantly antipsychotic-like behavioral profile (drug signature), with some secondary anxiolytic and antidepressant-like activity. C In vitro target profiling of ulotaront (SEP-363856). Additional in vitro and in vivo pharmacological studies showed that agonism at TAAR1 and 5-HT1A receptors contribute to the efficacy of ulotaront [32, 35]. D2 dopamine D2 receptor, 5-HT1A and 5-HT2A serotonin 1A and 2A receptor subtypes, ADHD attention deficit disorder with hyperactivity. *Antipsychotic (purple) and high-dose antipsychotic (dark purple); **antidepressant (green) and high-dose antidepressant (dark green)
Preclinical pharmacology
Functional profiling in vitro showed that ulotaront is a full agonist (Emax = 101%) at TAAR1 (EC50 = 0.14 µM). Ulotaront also exhibits binding to serotonin 5-HT1A receptors (Ki = 0.28 µM) where it acts as an agonist (EC50 = 2.3 µM; Emax = 75%). Subsequent mechanistic studies demonstrated that these receptor activities contribute to the effects of ulotaront in vivo (summarized below). In addition, ulotaront has affinity for serotonin 5-HT1D (Ki = 1.13 µM), 5-HT1B (Ki = 1.9 µM) and 5-HT7 (Ki = 0.03 µM) receptors, although only weak agonism was reported for 5-HT1B (EC50 = 15.6 µM; Emax = 22%) and 5-HT7 (EC50 = 6.7 µM; Emax = 41%) [32]. No appreciable binding, functional activity and/or in vivo receptor occupancy was seen at dopamine D2 or serotonin 5-HT2A receptors. For more details relating to the in vitro receptor profiling, we refer the reader to the original research article [32].
TAAR1 is a G-protein coupled receptor (GPRC) that is widely expressed in the rodent brain, albeit at very low levels. In rodents, receptor expression has been reported in monoaminergic nuclei including the ventral tegmental area (VTA), substantial nigra (SN), and dorsal raphe nucleus (DRN) as well as limbic brain regions (e.g., amygdala, hippocampus), basal ganglia, and the prefrontal cortex [36, 37]. Thus, it is not surprising that TAAR1 has been shown to affect dopaminergic, serotonergic and glutamatergic signaling and consequently modulate aspects of reward-processing, cognition and mood relevant to schizophrenia and other psychiatric disorders [38, 39]. Due to its low expression levels and lack of suitable, commercially-available tools such as antibodies for TAAR1, the synaptic and cellular localization of TAAR1 remains largely unexplored. Intracellular receptor localization has been reported, with evidence for plasma membrane expression following ligand-induced heterodimerization with other GPCRs [35, 37, 40,41,42,43,44,45,46].
TAAR1 agonists are broadly active in preclinical models/assays and have demonstrated antipsychotic, anti-addictive, pro-cognitive, antidepressant-like, and wake-promoting effects [35, 38, 39]. Ulotaront has been extensively studied in rodent models relevant to schizophrenia, supported by its antipsychotic-like profile in the in vivo phenotypic screening platform SmartCube® [32]. Efficacy has been reported in several additional models/assay, including stimulant-induced deficits/alterations in locomotor activity, prepulse inhibition, social interaction and cognition (Table 1). Importantly, some of the in vivo effects (including antipsychotic-like activity) were absent in TAAR1-knock out mice confirming the contribution of TAAR1 to ulotaront’s mechanism of action [35]. In addition, 5-HT1A receptors were shown to partially contribute towards the effects of ulotaront in the mouse PCP-induced hyperactivity assay. Although evidence exists for the therapeutic effects of 5-HT1A agonists in depression and anxiety-related disorders [47], the potential for efficacy of 5-HT1A agonists in schizophrenia, particularly in combination with TAAR1 agonism, remains to be explored.
The ability of TAAR1 to modulate dopaminergic circuits has attracted considerable interest in the context of schizophrenia and psychosis in general. VTA neuronal firing and electrically evoked dopamine release are reduced by TAAR1 full agonists, whereas generally opposite effects are seen with the TAAR1 antagonists such as N-(3-Ethoxy-phenyl)-4-pyrrolidin-1-yl-3-trifluoromethyl-benzamide (EPPTB) as well as in TAAR1-KO mice [48,49,50,51,52]. Interestingly, the inhibitory effects on dopaminergic neurotransmission appear to be most pronounced under hyperdopaminergic conditions. This is supported by recent findings, showing that ulotaront reduces the ketamine- induced increase in striatal dopamine synthesis capacity without producing an effect in naïve mice [53]. Elevated dopamine synthesis capacity has repeatedly been reported in schizophrenia patients and is not targeted by current antipsychotic treatments [54,55,56,57]. Whether ulotaront’s effects on dopamine synthesis capacity are mediated through direct action on midbrain dopaminergic neurons, or via upstream modulation of glutamatergic circuits, remains to be determined.
In contrast to some of the currently available antipsychotic drugs, and consistent with the lack of D2 receptor activity, neither selective TAAR1 agonists nor ulotaront induce catalepsy in rodents [32, 50]. Thus, TAAR1 agonists are unlikely to cause D2 antagonist-mediated extrapyramidal side effects (EPS, movement disorders), which constitute well-known side effect of the current class of antipsychotic drugs. Interestingly, TAAR1 agonists, including ulotaront, have also been reported to potentiate the antipsychotic properties of olanzapine and/or risperidone, highlighting their potential as adjunctive treatments to current antipsychotics [50, 59]. In addition, an increasing number of studies are implicating TAAR1 in the potential regulation of metabolic function and food reward behavior [38, 48, 62]. This could be of significant relevance considering that obesity, hyperglycemia, insulin resistance and dyslipidemia constitute major side effects of antipsychotic medication [63]. In contrast, TAAR1 agonists have been shown to decrease body weight in naïve rodents, prevent olanzapine-induced weight gain in rats, reduce food intake and excess body weight in diet-induced obese mice and attenuate binge-like eating in rats [43, 50, 58, 64]. Additional studies in mouse models of type 2 diabetes mellitus reported improved glucose tolerance and insulin sensitivity, as well as reduced plasma and liver triglyceride levels [42]. The underlying mechanisms may include TAAR1-mediated peripheral effects on glucose homeostasis and gastric emptying, and/or direct modulation of homeostatic and hedonic neurocircuits regulating energy balance. Thus, the current preclinical evidence suggests that TAAR1 agonists hold promise to improve several symptom domains of schizophrenia without causing motor impairments and metabolic dysregulation. In fact, the potential beneficial metabolic effects suggest that TAAR1 agonists may improve comorbid metabolic dysfunction in schizophrenia patients.
Lack of abuse liability in rodent models
A series of studies conducted in rodent models predictive of abuse potential in humans indicate that ulotaront is not likely to pose a risk of recreational abuse in humans [61]. Notably, single doses of ulotaront were associated with reductions in cocaine-primed induced reinstatement, and dose-dependently reduced cue-reinstated responding [61]. This is consistent with the effects of TAAR1 selective agonists which have been shown to inhibit the rewarding and reinforcing effects of drugs of abuse and drug-abuse related behaviors (reviewed in detail by Gainetdinov et al.; Pei et al.; Liu and Li) [38, 44, 65]. The mechanism is not fully elucidated but likely has its cellular and molecular basis in the attenuation of dopaminergic hyperactivity (i.e., accumulation of DA) induced by drugs of abuse [65].
Ulotaront: pharmacokinetics profile
The pharmacokinetic (PK) profile of ulotaront in preclinical species and humans has been well-characterized [66, 67]. Ulotaront is a small molecule with high solubility, high permeability, and low plasma protein binding in rodents and humans (unbound fraction, > 78%).
The ability of ulotaront to penetrate the blood–brain barrier has been demonstrated in mice and rats. Following single oral or intraperitoneal administration (10 mg/kg), ulotaront was rapidly absorbed and distributed to the brain with maximum concentrations reached within 30 min post dose in. The brain to plasma ratios (Cmax and AUC) were ≥ 4 and ≥ 2 in mice and rats, respectively [66].
In humans, ulotaront is well-absorbed after oral ingestion with a median time to maximum concentration (Tmax [90%-CI]) of 2.8 [1.0, 6.2] hours and a median effective terminal half-life (t½) of 7 [4.4, 11.4] hours. Daily dosing to steady state results in an accumulation ratio of 1.1, consistent with a once-daily dosing regimen. The PK profile of ulotaront exhibits a linear relationship between dose and plasma concentration across the presumed therapeutic dose range of 25–100 mg/day. However, the dose- and concentration–response (PK/PD) relationships for ulotaront in the acute (or maintenance) treatment of schizophrenia have not been established.
The metabolic and excretory pathways for ulotaront disposition have been well-characterized. Greater than 92% of ulotaront is excreted in urine as either parent drug (15%) or metabolites. A single major, inactive metabolite has been identified. There are no known clinically meaningful drug-drug interactions involving ulotaront or its metabolites against CYP enzymes or transporters.
Ulotaront: preliminary evidence for short-term efficacy
Ulotaront is currently being evaluated in a series of Phase 3 clinical trials designed to evaluate its short-term efficacy in the treatment of schizophrenia as well as its long-term effectiveness, safety, tolerability, and effect on measures of function and quality of life (Table 2). The dose selection for evaluation in schizophrenia patients was guided by an initial Phase 1 study in healthy male volunteers demonstrating robust rapid eye movement (REM) sleep-suppressing effects of ulotaront at 50 mg [68].
To date, the short-term efficacy of ulotaront in the treatment of schizophrenia has been evaluated in a Phase 2, multinational, 4-week, randomized, double-blind, parallel-group study of flexibly-dosed ulotaront (50–75 mg/day; n = 120) versus placebo (n = 125) in acutely psychotic adult inpatients with a DSM-5 diagnosis of schizophrenia [69]. The treatment sample was comprised of 64% males, mean age 30 years, with a mean PANSS total score at baseline of 101 (PANSS-positive and -negative subscale scores: 26 and 25, respectively), and a Brief Negative Symptom Scale (BNSS) total score of 37.
Significant improvement in the PANSS total score was observed at Week 4 (the primary efficacy endpoint; effect size, 0.45; Fig. 2), with separation from placebo observed as early as Week 3.

Significant improvement in PANSS total score during 4 weeks of treatment with ulotaront (50–75 mg/day) [69]
Statistically significant efficacy was also observed at Week 4 across all secondary efficacy measures (Table 3). Effect sizes were generally in the moderate range.
The efficacy of ulotaront on negative symptoms has been examined in more detail in a (pre-specified) analysis of four measures of negative symptoms (Fig. 3A) [70]. The ulotaront vs. placebo effect sizes at Week 4 for the PANSS-Marder negative symptom factor and BNSS total scores were similar (0.46 and 0.48, respectively) while the PANSS-negative subscale score effect size was somewhat lower (0.37).

Ulotaront (50–75 mg/day) efficacy in schizophrenia symptom domains in a randomized clinical trial. A Ulotaront was associated with significant improvement at Week 4 (effect size vs. placebo) across multiple measures of negative symptoms [70]. B Forest plot of endpoint effect sizes for UPSM-transformed PANSS factors: ulotaront vs. placebo [69]
However, it is well-known that change scores for PANSS-derived negative symptom measures are highly correlated with PANSS measures of positive symptoms. For example, the correlation between the PANSS-Marder positive and negative factor change scores has been shown to be 0.57 [71]. The correlation between PANSS Marder positive symptom factor and the BNSS total score is lower (0.36) but is still significant. These levels of correlation suggest that the improvement in negative symptoms that have been reported for many years for the D2 antipsychotic drugs may be attributed, in no small measure, to PANSS-positive symptom-related effects [71]. To address this cross-correlation issue, a pre-specified analysis of Week 4 improvement in negative symptoms on ulotaront was performed using an uncorrelated PANSS score matrix (UPSM) transformation of the two subdomains of the PANSS-negative symptom factor. The UPSM-transformed factors measure drug effects on PANSS symptom domains in schizophrenia with greater specificity by mathematically reducing the correlated improvements among individual PANSS items, resulting in low (0.04-to-0.10) between-factor correlations for UPSM-PANSS negative subfactors, apathy/avolition and deficit of expression (for a more detailed explanation, see: [71, 72]). The results of this analysis revealed positive effects for the UPSM negative symptom factors, apathy/avolition and deficit of expression (Fig. 3B) remaining over-and-above the expected improvements stemming from correlated effects on other PANSS factors.
To date, only an exploratory cross-study analysis is available based on a post-hoc enrichment strategy that compared endpoint effect sizes for ulotaront vs. placebo and pooled lurasidone vs. placebo on the UPSM-negative symptom factor [73]. In both treatment samples, enrichment was not based on identifying a subgroup with high baseline negative symptom severity. Instead, the enrichment strategy identified a subgroup of patients with low pre-randomization (Screen and Baseline) measurement heterogeneity on the Marder PANSS negative symptom (MPNS) construct. In the subgroup analysis of patients exhibiting high MPNS construct factor validity (with 69% of variance explained versus 37% for the non-enriched subgroup), treatment with ulotaront was associated with a notably larger effect size than lurasidone (0.84 vs. 0.33), suggesting that a prognostic enrichment strategy may be a more-efficient way to establish whether the clinical benefit of ulotaront (versus a D2-class MOA) extends to a specific improvement in negative symptoms [73]. Whether the TAAR1/5-HT1A MOA of ulotaront offers a differential efficacy advantage in the treatment of the negative symptom domain of schizophrenia when compared to D2 antipsychotic compounds awaits the results of ongoing Phase 3 clinical trials (Table 2).
Ulotaront: preliminary evidence for longer-term effectiveness
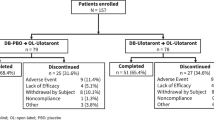
Of the 193 patients who completed the 4-week, double-blind, placebo-controlled trial of ulotaront, 157 patients (81.3%) continued into a 26-week open-label (OL) extension study, including 78 patients treated with ulotaront during the double-blind phase, and 79 patients treated with placebo (switching from placebo to ulotaront was accomplished while maintaining the initial study double-blind).
Twenty-six weeks of treatment with ulotaront was associated with continued improvement in symptoms of schizophrenia as measured by the PANSS total score (Fig. 4) [74]. In the group of patients who met responder criteria (≥ 30% reduction in PANSS total score) after completing double-blind treatment with ulotaront, the Kaplan–Meier estimate of the probability of relapse at the end of 26 weeks open-label treatment was 0.23.

Improvement in PANSS total score during 26 weeks of treatment with ulotaront (50–75 mg/day) [74]. PANSS positive and negative syndrome scale, DB double blind, OL open label, BL baseline
Continued improvement was also observed on the secondary efficacy measures such as the CGI-S score, the PANSS subscale scores, and the BNSS total score (Table 4). The great majority of patients met responder criteria of ≥ 30% reduction from DB baseline in the PANSS total score. A post-hoc analysis found that a large proportion of patients met stringent ≥ 50% reduction criteria at Week 26 (Table 4).
Ulotaront: preliminary safety and tolerability data
A preliminary safety and tolerability profile of ulotaront can be gleaned from the 4-week double-blind, placebo-controlled Phase 2 study [69], and the 26-week, open-label extension study [70]. As can be seen in Table 5, there were five AE terms that occurred (during the 4-week double-blind study) with an incidence ≥ 2% in the ulotaront group (and greater than placebo); all five AE terms had an incidence < 7% and the number needed to harm (NNH) value for each AE was > 50 for all but one AE (dyspepsia, NNH = 40), indicating that absolute risk increase (i.e., difference in event rate between drug and placebo) is likely to be of minimal clinical concern [75]. The combined incidence of extrapyramidal symptoms (akathisia, restlessness, musculoskeletal or joint stiffness, tremor, and nuchal rigidity) was 3.3% and 3.2% in the ulotaront and placebo groups respectively. Consistent with this favorable tolerability, the proportion of patients reporting any AE was lower on ulotaront compared to placebo (45.8% vs. 50.4%), the rate of AEs rated as “severe” was 5.8%, and the overall discontinuation rate for ulotaront was comparable to placebo (21.7% vs. 20.8%), with discontinuation due to an AE of 8.3% (vs. 6.4% on placebo; Table 5). Serous adverse events (SAEs) occurred in 2 patients treated with ulotaront versus 4 patients receiving placebo.
The 26-week, open-label extension study [74] provided additional evidence for a favorable benefit-risk ratio of ulotaront, most notably the overall 67% completion rate. As noted in the paper reporting the primary results of this study, [74] this completion rate compares favorably to completion rates at 24 weeks reported in the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study [76], which range from 39% for ziprasidone and quetiapine, to 55% for olanzapine.
On safety parameters, 26 weeks of treatment with ulotaront was associated with safety profile that was different from many of the currently approved antipsychotic medications. Most notably there were no clinically significant changes in median prolactin levels, mean weight, and median metabolic parameters (Table 5). Furthermore, standard movement disorder scales showed no clinically significant changes. For example, changes (mean (SD)) in the Simpson–Angus Scale mean score, Barnes Rating Scale for Drug-Induced Akathisia total score, and the Abnormal Involuntary Movement Scale total score were − 0.0 (0.1), − 0.1 (0.2), and 0.0 (0.1), respectively [74]. Worsening schizophrenia, headaches, insomnia, and anxiety were the only individual adverse events that occurred with an incidence ≥ 5% during 26 weeks of treatment with ulotaront (Table 5).
Overview of class effect differences between atypical antipsychotics and ulotaront
Atypical antipsychotic drugs whose MOA is mediated by antagonism at D2/5-HT2A receptors, exhibit a class-specific risk for certain adverse effects (e.g., extrapyramidal symptoms [EPS], cardiometabolic symptoms, hyperprolactinemia). As summarized in detail in recent reports [77] these D2-antipsychotic class-specific preferred terms have been empirically identified, utilizing an Empirical Bayes Geometric Mean (EBGM) disproportionality analysis of Food and Drug Administration Adverse Event Reporting System (FAERS) data, as any preferred term that meets the threefold greater EBGM threshold for drug vs. placebo. The application of the antipsychotic class-effect query has also been applied to historical trials of risperidone, lurasidone, olanzapine, and quetiapine [77, 78] to indicate that approximately half of the adverse events occurring in clinical trials of approved antipsychotic compounds are class effects. To illustrate this result in more detail, Fig. 5 displays the cumulative proportion of patients with schizophrenia having an AE on a second-generation antipsychotic drug at-or-above the threefold disproportional EBGM. As can be seen, there is a notable difference in the cumulative proportion of antipsychotic class-specific AEs for ulotaront when compared to lurasidone, olanzapine, and quetiapine [75].

Second generation class-related D2 antipsychotic adverse events based on FAERS data (n = 11 antipsychotics). The x-axis is the fold-increase in disproportional reporting of each AE. The y-axis is the cumulative proportion of patients with class-specific AEs that meet the threefold EBGM threshold [77]. EBGM Empirical Bayes Geometric Mean, FAERS Food and Drug Administration Adverse Event Reporting System (FAERS) data
Conclusion
Ulotaront is the first TAAR1 agonist that has progressed to Phase 3 clinical trials for the treatment of schizophrenia. Phase 2 clinical data suggest that ulotaront may treat a spectrum of symptoms associated with schizophrenia, including both positive and negative symptoms. In addition, clinical data point to a safety and tolerability profile distinct from the SGA and FGA antipsychotic classes, consistent with the absence of D2 receptor blockade. Though the MOA of ulotaront in the treatment of schizophrenia has not been fully elucidated, a growing body of evidence [32, 35] suggests that the efficacy of ulotaront is mediated through agonism at TAAR1 and serotonin 5-HT1A receptors. Via this novel mechanism, it is thought that ulotaront may provide a distinct risk/benefit profile notably lacking D2 antipsychotic class-related AEs (e.g., EPS, hyperprolactinemia, and adverse weight and metabolic effects). Emerging Phase 3 clinical data from this compound will not only be fundamental to our understanding of ulotaront but may help elucidate the therapeutic utility of TAAR1 agonists for the treatment of schizophrenia and beyond [79,80,81,82,83].
References
Kane JM (2022) A new treatment paradigm: targeting trace amine-associated receptor 1 (TAAR1) in schizophrenia. J Clin Psychopharmacol 42(5 Suppl 1):S1–S13. https://doi.org/10.1097/JCP.0000000000001596
Kahn RS, Sommer IE, Murray RM et al (2015) Schizophrenia. Nat Rev Dis Primers 12(1):15067
Díaz-Caneja CM, Pina-Camacho L, Rodríguez-Quiroga A, Fraguas D, Parellada M, Arango C (2015) Predictors of outcome in early-onset psychosis: a systematic review. NPJ Schizophr 4(1):14005
Correll CU, Abi-Dargham A, Howes O (2022) Emerging treatments in schizophrenia. J Clin Psychiatry 83(1):SU21204IPI. https://doi.org/10.4088/JCP.SU21204IP1
GBD 2017 Disease and Injury Incidence and Prevalence Collaborators (2018) Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392:1789–1858. https://doi.org/10.1016/S0140-6736(18)32279-7
Chong HY, Teoh SL, Wu DB-C et al (2016) Global economic burden of schizophrenia: a systematic review. Neuropsychiatr Dis Treat 12:357–373
American Psychiatric Association (2020) The American Psychiatric Association practice guideline for the treatment of patients with schizophrenia, 3rd edn. American Psychiatric Association Publishing. https://doi.org/10.1176/appi.books.9780890424841
Stępnicki P, Kondej M, Kaczor AA (2018) Current concepts and treatments of schizophrenia. Molecules 23(8):2087
Kantrowitz JT (2020) Targeting serotonin 5-HT2A receptors to better treat schizophrenia: rationale and current approaches. CNS Drugs 34(9):947–959
Leucht S, Cipriani A, Spineli L et al (2013) Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet 382(9896):951–962
Correll CU, Solmi M, Croatto G et al (2022) Mortality in people with schizophrenia: a systematic review and meta-analysis of relative risk and aggravating or attenuating factors. World Psychiatry 21(2):248–271
Tanskanen A, Tiihonen J, Taipale H (2018) Mortality in schizophrenia: 30-year nationwide follow-up study. Acta Psychol Scand 138:492–499
Drosos P, Brønnick K, Joa I et al (2020) One-year outcome and adherence to pharmacological guidelines in first-episode schizophrenia: results from a consecutive cohort study. J Clin Psychopharmacol 40(6):534–540
Shimomura Y, Kikuchi Y, Suzuki T, Uchida H, Mimura M, Takeuchi H (2020) Antipsychotic treatment in the maintenance phase of schizophrenia: an updated systematic review of the guidelines and algorithms. Schizophr Res 215:8–16
Hiwot S, Henock A (2018) A study to assess the prevalence of antipsychotics non-adherence and its associated factors among patients with schizophrenia in. JOJ Nurse Health Care 6:001–005
Kaplan G, Casoy J, Zummo J (2013) Impact of long-acting injectable antipsychotics on medication adherence and clinical, functional, and economic outcomes of schizophrenia. Patient Prefer Adherence 7:1171–1180
Fusar-Poli P, Papanastasiou E, Stahl D et al (2015) Treatments of negative symptoms in schizophrenia: meta-analysis of 168 randomized placebo-controlled trials. Schizophr Bull 41(4):892–899
Aleman A, Lincoln TM, Bruggeman R et al (2017) Treatment of negative symptoms: where do we stand, and where do we go? Schizophr Res 186:55–62
Milev P, Ho BC, Arndt S, Andreasen NC (2005) Predictive values of neurocognition and negative symptoms on functional outcome in schizophrenia: a longitudinal first-episode study with 7-year follow-up. Am J Psychiatry 162(3):495–506. https://doi.org/10.1176/appi.ajp.162.3.495
Carbon M, Correll CU (2014) Thinking and acting beyond the positive: the role of the cognitive and negative symptoms in schizophrenia. CNS Spectr 19(Suppl 1):38–52. https://doi.org/10.1017/S1092852914000601. (quiz 35–37, 53)
Rabinowitz J, Berardo CG, Bugarski-Kirola D, Marder S (2013) Association of prominent positive and prominent negative symptoms and functional health, well-being, healthcare-related quality of life and family burden: a CATIE analysis. Schizophr Res 150(2–3):339–342
Harvey PD, Strassnig M (2012) Predicting the severity of everyday functional disability in people with schizophrenia: cognitive deficits, functional capacity, symptoms, and health status. World Psychiatry 11(2):73–79. https://doi.org/10.1016/j.wpsyc.2012.05.004
Mucci A, Galderisi S, Gibertoni D, Rossi A, Rocca P, Bertolino A et al (2021) Factors associated with real-life functioning in persons with schizophrenia in a 4-year follow-up study of the italian network for research on psychoses. JAMA Psychiat 78:550–559
Rabinowitz J, Werbeloff N, Caers I et al (2013) Negative symptoms in schizophrenia—the remarkable impact of inclusion definitions in clinical trials and their consequences. Schizophr Res 150(2–3):334–338
Mäkinen J, Miettunen J, Isohanni M, Koponen H (2008) Negative symptoms in schizophrenia: a review. Nord J Psychiatry 62(5):334–341
McCleery A, Nuechterlein KH (2019) Cognitive impairment in psychotic illness: prevalence, profile of impairment, developmental course, and treatment considerations. Dialogues Clin Neurosci 21(3):239–248. https://doi.org/10.31887/DCNS.2019.21.3/amccleery
Sheffield JM, Karcher NR, Barch DM (2018) Cognitive deficits in psychotic disorders: a lifespan perspective. Neuropsychol Rev 28(4):509–533
Halff EF, Rutigliano G, Garcia-Hidalgo A, Howes OD (2022) Trace amine-associated receptor 1 (TAAR1) agonism as a new treatment strategy for schizophrenia and related disorders. Trends Neurosci. https://doi.org/10.1016/j.tins.2022.10.010
Correll CU, Schooler NR (2020) Negative symptoms in schizophrenia: a review and clinical guide for recognition, assessment, and treatment. Neuropsychiatr Dis Treat 2020(16):519–534. https://doi.org/10.2147/NDT.S225643.eCollection
Hopkins SC, Lew R, Zeni C, Koblan KS (2022) Challenges in the clinical development of non-D2 compounds for schizophrenia. Curr Med Res Opin 25:1–5. https://doi.org/10.1080/03007995.2022.2147342
Fleischhacker WW, Podhorna J, Gröschl M et al (2021) Efficacy and safety of the novel glycine transporter inhibitor BI 425809 once daily in patients with schizophrenia: a double-blind, randomised, placebo-controlled phase 2 study. Lancet Psychiatry 8(3):191–201
Dedic N, Jones PG, Hopkins SC et al (2019) SEP-363856, a novel psychotropic agent with a unique, non-D2 receptor mechanism of action. J Pharmacol Exp Ther 371(1):1–14
Vincent F, Nueda A, Lee J, Schenone M, Prunotto M, Mercola M (2022) Phenotypic drug discovery: recent successes, lessons learned and new directions. Nat Rev Drug Discov 21(12):899–914
Leahy E, Varney M, Brunner D (2020) Use of phenotypic screening in mice in the development of a novel non-D2-receptor-targeting drug for the treatment of schizophrenia. In: Isherwood B, Augustin A (eds) Phenotypic drug discovery. Drug Discovery eBook Collection. https://doi.org/10.1039/9781839160721-00175
Saarinen M, Mantas I, Flais I et al (2022) TAAR1 dependent and independent actions of the potential antipsychotic and dual TAAR1/5-HT1A receptor agonist SEP-383856. Neuropsychopharmacology 47:2319–2329
Borowsky B, Adham N, Jones KA et al (2001) Trace amines: identification of a family of mammalian G protein-coupled receptors. Proc Natl Acad Sci U S A 98(16):8966–8971. https://doi.org/10.1073/pnas.151105198
Bunzow JR, Sonders MS, Arttamangkul S et al (2001) Amphetamine, 3,4-methylenedioxymethamphetamine, lysergic acid diethylamide, and metabolites of the catecholamine neurotransmitters are agonists of a rat trace amine receptor. Mol Pharmacol 60(6):1181–1188. https://doi.org/10.1124/mol.60.6.1181
Gainetdinov RR, Hoener MC, Berry MD (2018) Trace amines and their receptors. Pharmacol Rev 70(3):549–620
Dedic N, Dworak H, Zeni C, Rutigliano G, Howes OD (2021) Therapeutic potential of TAAR1 agonists in schizophrenia: evidence from preclinical models and clinical studies. Int J Mol Sci 22(24):13185. https://doi.org/10.3390/ijms222413185
Lindemann L, Hoener MC (2005) A renaissance in trace amines inspired by a novel GPCR family. Trends Pharmacol Sci 26(5):274–281. https://doi.org/10.1016/j.tips.2005.03.007
Revel FG, Moreau JL, Pouzet B et al (2013) A new perspective for schizophrenia: TAAR1 agonists reveal antipsychotic- and antidepressant-like activity, improve cognition and control body weight. Mol Psychiatry 18(5):543–556. https://doi.org/10.1038/mp.2012.57
Raab S, Wang H, Uhles S et al (2015) Incretin-like effects of small molecule trace amine-associated receptor 1 agonists. Mol Metab 5(1):47–56. https://doi.org/10.1016/j.molmet.2015.09.015
Miller GM, Verrico CD, Jassen A et al (2005) Primate trace amine receptor 1 modulation by the dopamine transporter. J Pharmacol Exp Ther 313(3):983–994. https://doi.org/10.1124/jpet.105.084459
Pei Y, Asif-Malik A, Canales JJ (2016) Trace amines and the trace amine-associated receptor 1: pharmacology, neurochemistry, and clinical implications. Front Neurosci 5(10):148. https://doi.org/10.3389/fnins.2016.00148
Harmeier A, Obermueller S, Meyer CA et al (2015) Trace amine-associated receptor 1 activation silences GSK3β signaling of TAAR1 and D2R heteromers. Eur Neuropsychopharmacol 25(11):2049–2061. https://doi.org/10.1016/j.euroneuro.2015.08.011
Espinoza S, Salahpour A, Masri B et al (2011) Functional interaction between trace amine-associated receptor 1 and dopamine D2 receptor. Mol Pharmacol 80(3):416–425. https://doi.org/10.1124/mol.111.073304
Celada P, Bortolozzi A, Artigas F (2013) Serotonin 5-HT1A receptors as targets for agents to treat psychiatric disorders: rationale and current status of research. CNS Drugs 27(9):703–716
Rutigliano G, Zucchi R (2020) Molecular variants in human trace amine-associated receptors and their implications in mental and metabolic disorders. Cell Mol Neurobiol 40(2):239–255. https://doi.org/10.1007/s10571-019-00743-y
Revel FG, Moreau JL, Gainetdinov RR et al (2011) TAAR1 activation modulates monoaminergic neurotransmission, preventing hyperdopaminergic and hypoglutamatergic activity. Proc Natl Acad Sci U S A 108(20):8485–8490. https://doi.org/10.1073/pnas.1103029108
Revel FG, Moreau JL, Gainetdinov RR et al (2012) Trace amine-associated receptor 1 partial agonism reveals novel paradigm for neuropsychiatric therapeutics. Biol Psychiatry 72(11):934–942. https://doi.org/10.1016/j.biopsych.2012.05.014
Leo D, Mus L, Espinoza S, Hoener MC, Sotnikova TD, Gainetdinov RR (2014) Taar1-mediated modulation of presynaptic dopaminergic neurotransmission: role of D2 dopamine autoreceptors. Neuropharmacology 81:283–291. https://doi.org/10.1016/j.neuropharm.2014.02.007
Bradaia A, Trube G, Stalder H et al (2009) The selective antagonist EPPTB reveals TAAR1-mediated regulatory mechanisms in dopaminergic neurons of the mesolimbic system. Proc Natl Acad Sci U S A 106(47):20081–20086. https://doi.org/10.1073/pnas.0906522106
Kokkinou M, Irvine EE, Bonsall DR et al (2021) Reproducing the dopamine pathophysiology of schizophrenia and approaches to ameliorate it: a translational imaging study with ketamine. Mol Psychiatry 26:2562–2576
Kim E, Howes OD, Veronese M et al (2017) Presynaptic dopamine capacity in patients with treatment-resistant schizophrenia taking clozapine: an [18F]DOPA PET study. Neuropsychopharmacology 42(4):941–950. https://doi.org/10.1038/npp.2016.258
McCutcheon R, Beck K, Jauhar S, Howes OD (2018) Defining the locus of dopaminergic dysfunction in schizophrenia: a meta-analysis and test of the mesolimbic hypothesis. Schizophr Bull 44(6):1301–1311. https://doi.org/10.1093/schbul/sbx180
Howes OD, Kambeitz J, Kim E et al (2012) The nature of dopamine dysfunction in schizophrenia and what this means for treatment. Arch Gen Psychiatry 69(8):776–786. https://doi.org/10.1001/archgenpsychiatry.2012.169
Jauhar S, Nour MM, Veronese M et al (2017) A test of the transdiagnostic dopamine hypothesis of psychosis using positron emission tomographic imaging in bipolar affective disorder and schizophrenia. JAMA Psychiat 74(12):1206–1213. https://doi.org/10.1001/jamapsychiatry.2017.2943
Begni V, Sanson A, Luoni A et al (2021) Towards novel treatments for schizophrenia: molecular and behavioural signatures of the psychotropic Agent SEP-363856. Int J Mol Sci 22(8):4119
Liang L, Ren X, Xu J, Ma Y, Xue Y, Zhuang T, Zhang G (2022) Effect of co-treatment of olanzapine with SEP-363856 in mice models of schizophrenia. Molecules 27(8):2550. https://doi.org/10.3390/molecules27082550
Ren X, Xiong J, Liang L, Chen Y, Zhang G (2022) Potential antidepressant action of duloxetine co-Administered with the TAAR1 receptor agonist SEP-363856 in mice. Molecules 27(9):2755. https://doi.org/10.3390/molecules27092755
Synan C, Bowen C, Heal DJ et al (2022) Ulotaront, a novel TAAR1 agonist with 5-HT1A agonist activity, lacks abuse liability and attenuates cocaine cue-induced relapse in rats. Drug Alcohol Depend 231:109261. https://doi.org/10.1016/j.drugalcdep.2021.109261
Moore CF, Sabino V, Cottone P (2018) Trace amine associated receptor 1 (TAAR1) modulation of food reward. Front Pharmacol 27(9):129. https://doi.org/10.3389/fphar.2018.00129
Pillinger T, McCutcheon RA, Vano L et al (2020) Comparative effects of 18 antipsychotics on metabolic function in patients with schizophrenia, predictors of metabolic dysregulation, and association with psychopathology: a systematic review and network meta-analysis. Lancet Psychiatry 7(1):64–77. https://doi.org/10.1016/S2215-0366(19)30416-X
Ferragud A, Howell AD, Moore CF et al (2017) The trace amine-associated receptor 1 agonist RO5256390 blocks compulsive, binge-like eating in rats. Neuropsychopharmacology 42(7):1458–1470. https://doi.org/10.1038/npp.2016.233
Liu JF, Li JX (2018) TAAR1 in addiction: looking beyond the tip of the iceberg. Front Pharmacol 27(9):279. https://doi.org/10.3389/fphar.2018.00279.PMID:29636691;PMCID:PMC5881156
Galluppi GR, Polhamus DG, Fisher JM, Hopkins SC, Koblan KS (2021) Population pharmacokinetic analysis of ulotaront in subjects with schizophrenia. CPT Pharmacometr Syst Pharmacol 10:1245–1254
Xiao G, Chen YL, Dedic N, Xie L, Koblan KS, Galluppi GR (2022) In vitro ADME and preclinical pharmacokinetics of ulotaront, a TAAR1/5-HT1A receptor agonist for the treatment of schizophrenia. Pharm Res 39:837–850
Hopkins SC, Dedic N, Koblan KS (2021) Effect of TAAR1/5-HT1A agonist SEP-363856 on REM sleep in humans. Transl Psychiatry 11(1):228
Koblan KS, Kent J, Hopkins SC, Krystal JH, Cheng H, Goldman R, Loebel A (2020) A non-D2 receptor binding drug for the treatment of schizophrenia. N Engl J Med 382:1497–1506
Dworak H, Hopkins SC, Koblan KS et al (2021) Effects of SEP-363856, a novel TAAR1 agonist, on negative symptoms in schizophrenia: results across an initial double-blind acute study, and a 6-month, open-label extension study. In: Poster presentation at the American Psychiatric Association annual meeting, May 1–5, 2021
Hopkins SC, Ogirala A, Loebel A, Koblan KS (2018) Transformed PANSS factors intended to reduce pseudospecificity among symptom domains and enhance understanding of symptom change in antipsychotic-treated patients with schizophrenia. Schizophr Bull 44:593–602
Hopkins SC, Ogirala A, Loebel A, Koblan KS (2020) Characterization of specific and distinct patient types in clinical trials of acute schizophrenia using an uncorrelated PANSS score matrix transform (UPSM). Psychiatry Res 294:113569. https://doi.org/10.1016/j.psychres.2020.113569
Hopkins SC, Tomioka S, Koblan KS (2022) A general theory of construct enrichment: inclusion criteria for symptom structure not severity. In: Poster presentation at the International Society for CNS Clinical Trials and Methodology annual meeting, Boston
Correll CU, Koblan KS, Hopkins SC et al (2021) Safety and effectiveness of ulotaront (SEP-363856) in schizophrenia: results of a 6-month, open-label extension study. NPJ Schizophr 7:63. https://doi.org/10.1038/s41537-021-00190-z.xxxxx
Citrome L, Ketter TA (2013) When does a difference make a difference? Interpretation of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Int J Clin Pract 67:407–411
Lieberman JA, Stroup TS, McEvoy JP et al (2005) Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 353:1209–1223
Hopkins SC, Ogirala A, Worden M, Koblan KS (2021) Depicting safety profile of TAAR1 agonist ulotaront relative to reactions anticipated for a dopamine D2-based pharmacological class in FAERS. Clin Drug Investig 41(12):1067–1073
Hopkins SC, Ogirala A, Zeni C, Worden M, Koblan KS (2022) Depicting risperidone safety profiles in clinical trials across different diagnoses using a dopamine D2-based pharmacological class effect query defined by FAERS. Clin Drug Investig 42(12):1113–1121. https://doi.org/10.1007/s40261-022-01218-7. (Epub 2022 Nov 9)
NCT04072354. A clinical trial to study the efficacy and safety of an investigational drug in acutely psychotic people with schizophrenia. https://clinicaltrials.gov/ct2/show/NCT04072354?term=NCT04072354&draw=2&rank=1. Accessed 7 Nov 2022
NCT04092686. A clinical trial that will study the efficacy and safety of an investigational drug in acutely psychotic people with schizophrenia. https://clinicaltrials.gov/ct2/show/NCT04092686?term=NCT04092686&draw=2&rank=1. Accessed 7 Nov 2022
NCT04109950. A clinical study to evaluate the long-term safety and tolerability of an investigational drug in people with schizophrenia. https://clinicaltrials.gov/ct2/show/NCT04109950?term=NCT04109950&draw=2&rank=1. Accessed 7 Nov 2022
NCT04115319. A study of the long-term safety and tolerability of an investigational drug in people with schizophrenia. https://clinicaltrials.gov/ct2/show/NCT04115319?term=Sep-363856&cond=Schizophrenia&draw=4&rank=15. Accessed 7 Nov 2022
NCT05628103. A clinical study that will evaluate how well SEP-363856 works and how safe it is in people with schizophrenia that switch to SEP-363856 from their current antipsychotic medication. https://clinicaltrials.gov/ct2/show/record/NCT05628103?term=sep-363856+switch&draw=2&rank=1. Accessed 5 Jan 2023
Acknowledgements
Sunovion discovered ulotaront in collaboration with PsychoGenics based in part on a mechanism-independent approach using the in vivo phenotypic SmartCube® platform and associated artificial intelligence algorithms. Supported by funding from Sunovion Pharmaceuticals Inc. and Otsuka Pharmaceuticals Development & Commercialization, Inc. Medical writing support was provided by Edward Schweizer, MD of Paladin Consulting Group, and was funded by Sunovion Pharmaceuticals Inc. Sunovion discovered Ulotaront in collaboration with PsychoGenics based in part on a mechanism-independent approach using the in vivo phenotypic SmartCube® platform and associated artificial intelligence algorithms.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Achtyes has served on advisory boards or consulted for Alkermes, Atheneum, Janssen, Karuna, Lundbeck/Otsuka, Roche, Sunovion and Teva. He has received research support from Alkermes, Astellas, Biogen, Boehringer-Ingelheim, InnateVR, Janssen, National Network of Depression Centers, Neurocrine Biosciences, Novartis, Otsuka, Pear Therapeutics, and Takeda. He serves as an advisor to CAPNOS Zero, the World Psychiatric Association and Clubhouse International, and the SMI Adviser LAI Center of Excellence (all unpaid). Drs. Hopkins, Dedic, Dworak, Zeni, and Koblan are employees of Sunovion Pharmaceuticals Inc.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Achtyes, E.D., Hopkins, S.C., Dedic, N. et al. Ulotaront: review of preliminary evidence for the efficacy and safety of a TAAR1 agonist in schizophrenia. Eur Arch Psychiatry Clin Neurosci 273, 1543–1556 (2023). https://doi.org/10.1007/s00406-023-01580-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00406-023-01580-3