
Abstract
The role of microglial cells in the pathogenesis of Alzheimer’s disease (AD) neurodegeneration is unknown. Although several works suggest that chronic neuroinflammation caused by activated microglia contributes to neurofibrillary degeneration, anti-inflammatory drugs do not prevent or reverse neuronal tau pathology. This raises the question if indeed microglial activation occurs in the human brain at sites of neurofibrillary degeneration. In view of the recent work demonstrating presence of dystrophic (senescent) microglia in aged human brain, the purpose of this study was to investigate microglial cells in situ and at high resolution in the immediate vicinity of tau-positive structures in order to determine conclusively whether degenerating neuronal structures are associated with activated or with dystrophic microglia. We used a newly optimized immunohistochemical method for visualizing microglial cells in human archival brain together with Braak staging of neurofibrillary pathology to ascertain the morphology of microglia in the vicinity of tau-positive structures. We now report histopathological findings from 19 humans covering the spectrum from none to severe AD pathology, including patients with Down’s syndrome, showing that degenerating neuronal structures positive for tau (neuropil threads, neurofibrillary tangles, neuritic plaques) are invariably colocalized with severely dystrophic (fragmented) rather than with activated microglial cells. Using Braak staging of Alzheimer neuropathology we demonstrate that microglial dystrophy precedes the spread of tau pathology. Deposits of amyloid-beta protein (Aβ) devoid of tau-positive structures were found to be colocalized with non-activated, ramified microglia, suggesting that Aβ does not trigger microglial activation. Our findings also indicate that when microglial activation does occur in the absence of an identifiable acute central nervous system insult, it is likely to be the result of systemic infectious disease. The findings reported here strongly argue against the hypothesis that neuroinflammatory changes contribute to AD dementia. Instead, they offer an alternative hypothesis of AD pathogenesis that takes into consideration: (1) the notion that microglia are neuron-supporting cells and neuroprotective; (2) the fact that development of non-familial, sporadic AD is inextricably linked to aging. They support the idea that progressive, aging-related microglial degeneration and loss of microglial neuroprotection rather than induction of microglial activation contributes to the onset of sporadic Alzheimer’s disease. The results have far-reaching implications in terms of reevaluating current treatment approaches towards AD.
Similar content being viewed by others


Introduction
Microglia are ubiquitously distributed cells in the central nervous system (CNS) and recognized to be involved in innate immunity and surveillance of the parenchyma [12, 15, 18, 33, 34]. Following acute insults to the CNS, microglial cells become activated and participate in the natural wound healing response [22, 45]. Their role in the pathogenesis of Alzheimer’s disease (AD) remains controversial, as they have been implicated in both neuroprotective and neurotoxic ways [11, 15, 45]. A potential involvement of microglia is most often seen within the context of the amyloid cascade hypothesis [16] where it is thought that fibrillar Aβ deposits constitute a chronic inflammatory stimulus triggering long-lasting microglial activation that results in the production of substances with neurotoxic activities, which contribute to the onset of neurodegeneration [11, 28, 32]. There are, however, caveats associated with the amyloid cascade/neuroinflammation theory of AD perhaps most importantly the fact that clear benefits of non-steroidal anti-inflammatory drugs for delaying onset or reversing cognitive dysfunction in Alzheimer’s patients have not been demonstrated [2, 26]. A direct pathogenic link between amyloid plaques and neurodegeneration also continues to remain elusive, because Aβ deposits and neurofibrillary tangles do not arise in the same anatomical locations, and cognitive impairment does not correlate with amyloid plaque load but with presence of neurofibrillar pathology evident as tau-positive structures such as neuropil threads, neurofibrillary tangles, and neuritic plaques [4, 7, 13, 37]. Additionally, unequivocal identification of activated microglia in human brain has been difficult to achieve since there is no single biomolecular marker for distinguishing activated cells from non-activated ones, and this has been complicated further by the fact that many microglial cells in the aged human brain are dystrophic showing morphological features indicative of senescence and/or degeneration rather than of activation, such as fragmented cytoplasmic processes [44]. However, recognition of dystrophic microglia has provided a new perspective on the potential involvement of microglia in aging-related neurodegenerative diseases, namely, that neurodegeneration may be occurring secondarily to microglial senescence and associated loss of microglial neuroprotection [43, 45]. This microglial dysfunction hypothesis is worth considering because it (1) is based on the premise that microglia, like other glial cells, are basically neuron-supporting cells and thus neuroprotective; (2) is compatible with the fact that the incidence of AD increases inevitably with aging and therefore considers primarily non-familial, sporadic forms of the disease, which represent greater than 90% of all cases. The goal of the current study was to investigate directly the microglial dysfunction hypothesis by examining microglial cells in human brain at high resolution in the immediate vicinity of tau-positive structures in hopes of being able to determine conclusively whether degenerating neuronal structures are associated with activated or with dystrophic microglia. The implications of resolving this issue are significant because if indeed there were evidence for microglial degeneration, understanding of AD pathogenesis would be steered into a new direction that would produce fundamentally different treatment approaches: rather than treating AD patients with anti-inflammatory drugs to deactivate microglia therapeutic strategies aimed at preserving or enhancing microglial cell function could prove to be more effective.
Materials and methods
All human brain samples for light microscopy were obtained from the brain bank at the Institute for Clinical Neuroanatomy, University of Frankfurt (Table 1). No patient data other than age, sex, and diagnosis were disclosed. All brains were fixed and stored by immersion in a 4% aqueous solution of formaldehyde for variable lengths of time beginning at the time of brain removal from the skull. Blocks containing the temporal lobe were embedded in polyethylene glycol (PEG 1000, Merck), and sections were cut on a macrotome at 100 μm in the frontal plane at the level of the uncus. Sections were processed for immunohistochemical staining using primary antibodies, ionized calcium binding adaptor molecule 1 (iba1) (rabbit polyclonal, 1:1,000, Wako) for detecting microglia, AT8 (mouse monoclonal, 1:2,000, Pierce Endogen) for detecting human PHF-tau, and 4G8 (mouse monoclonal, 1:4,000, Calbiochem) for detecting Aβ. Primary antibody binding sites were visualized using biotinylated secondary anti-mouse or anti-rabbit antibodies made in goat (Vector, 1:200) followed by streptavidin-peroxidase. Peroxidase substrates were diaminobenzidine (DAB-H2O2) and Vector SG substrate (Cat. No. SK-4700), to yield brown and blue-gray reaction products, respectively. For double immunohistochemical staining, the DAB substrate reaction was always performed first and sections were rinsed thoroughly in phosphate buffered saline, pH 7.2–7.4, before proceeding with the second immunostaining reaction. Negative controls consisted of omitting either the primary antibodies or incubating primary antibodies with mismatched secondary antibodies (e.g. primary mouse with biotinylated goat-anti-rabbit). In addition to immunohistochemistry, Aβ was visualized also using the Campbell–Switzer advanced silver technique and neurofibrillary degeneration was shown using the Gallyas technique, as described previously [5]. Preparations were examined and photographed using an Olympus Vanox microscope, as well as a Zeiss Axioplan microscope equipped with differential interference optics (Fig. 7). Three-dimensional (3-D) reconstruction was performed using analySIS Soft imaging system (Olympus).
Results
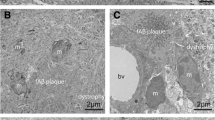
As a first step in our investigation it was essential to have a reliable histological method for demonstrating all microglial cells in archival human brain specimens stored for extended periods of time in formalin. To date no such method has been described, and published procedures demonstrate only partial visualization of microglial cells, due to loss or masking of antibody binding sites as a result of prolonged fixation [27]. In initial screening studies using 100-μm-thick sections of formalin-fixed human brains, we found that an antibody against the iba1 [19] provided excellent visualization of ramified microglia in normal, non-diseased control subjects (Fig. 1). Unlike antibodies against ferritin or HLA-DR antigens previously used for staining microglia in human brain, anti-iba 1 worked consistently well and labeled the entire microglial population, as judged by the great density of cells revealed throughout coronal sections of the temporal lobe (Fig. 1a, b). No special pretreatment of sections for antigen unmasking was required. Anti-iba1 staining also facilitated differentiating between resting and activated microglia using cell hypertrophy as the distinguishing feature (Fig. 1c, d). Negative control sections with the primary antibody omitted showed no staining at all. With this method, we set out to first study microglial morphology in cases of Alzheimer’s disease showing the classic pathology of senile plaques and neurofibrillary tangles (Braak stages V and VI, case nos. 10–13; Table 1). By combining anti-iba1 staining with anti-tau immunolabeling (AT8 antibody) we were able to generate clear representations of microglial cells and degenerating neuronal structures (Fig. 2). The extent of neurodegeneration was visible even to the naked eye on AT8-stained coronal sections encompassing the temporal lobe up to the middle temporal gyrus at the level of the uncus. Microscopically, anti-tau immunolabeling, as well as silver staining using the Gallyas method, prominently highlighted neurofibrillary tangles, neuritic plaques, and neuropil threads, which were readily distinguished from microglial cells in these double-stained preparations (Fig. 2a–d). Negative controls with mismatched secondary antibodies showed no staining at all. The visualization of microglia with anti-iba1 failed to show any evidence of microglial activation, such as hypertrophy of the cells’ processes or increased cell density due to proliferation. However, clusters of microglia which have been described consistently in the literature were abundant and always associated with neuritic plaques. On high magnification, the structure of microglial cells was strikingly abnormal in that the cytoplasm of nearly all cells was fragmented into many small pieces (Fig. 2). The use of 100-μm-thick sections allowed for 3-D analysis of microglial cell structure by taking photomicrographs in multiple focal planes (Fig. 2e) and then digitally reassembling the z-stack of four or more images into a composite final micrograph (Fig. 2f). This type of 3-D reconstruction clearly established that microglial fragmentation (cytorrhexis) was not due to selective focusing but instead represented the actual morphology of both clustered and surrounding cells. We therefore concluded that neurodegenerative changes of AD were associated not with microglial activation but with microglial fragmentation, suggesting ongoing degeneration. To further substantiate this idea we sought to corroborate an association of microglial fragmentation and tau pathology in other cases at extreme ends of the spectrum of neurofibrillary pathology, namely, in young individuals with minimal tau pathology and in subjects with Down’s syndrome (case nos. 5, 14, 15; Table 1) using the same double-staining and 3-D reconstruction procedures as for the AD cases. We were able to determine that an area of minimal tau pathology in the hippocampus of a young subject (case no. 5) was accompanied by selective microglial cytorrhexis, whereas a non-pathological young control subject (case no. 1) revealed intact ramified microglial cells in the same brain region (Fig. 3). On the opposite end of the spectrum were two cases of Down’s syndrome both of which demonstrated widespread neurofibrillary and amyloid pathology virtually identical to the most advanced forms of AD. As shown in Fig. 4, these subjects revealed complete destruction and total loss of microglial cell integrity to the point where hardly a single intact microglial cell could be identified anywhere in the section. Iba1-stained sections of both Down’s subjects were littered with microglial debris consisting of numerous cell fragments, spheroids, and severely atrophied and disfigured microglia. These findings clearly confirmed our notion that microglial degeneration and neurodegeneration progress in synchrony.

Anti-iba1 immunohistochemistry produces specific staining of microglia in archival human brain tissue. a, b Low power views demonstrate abundance and even spacing of microglial cells throughout the cortical gray matter of the entorhinal cortex in a non-pathological control subject (case no. 3; Table 1). Pial surface is on far left in a. All cells show a non-activated, non-dystrophic morphology typical for normal, resting microglia. c, d Comparison of resting and activated microglial morphology in two age-matched individuals (case nos. 2 and 6, respectively). Activated microglia (d) show cytoplasmic hypertrophy in a subject with sepsis. Scale bars 500 μm (a), 100 μm (b), 50 μm (c, d)

Microglial fragmentation is widespread in Alzheimer’s disease brain and colocalized with tau pathology. Double-label immunohistochemistry for microglia (iba1) and tau (AT8) in the entorhinal cortex (EC) of case no. 12 (a–c). a Low power view reveals tau-positive neuritic plaques (black spots) in the cortical gray matter and diffusely distributed neuropil threads and tangles (lower right); microglia (brown) are evenly distributed throughout. Higher magnification shows neurofibrillary tangle and neuropil threads (b) and neuritic plaque (c), which are surrounded by fragmented microglia lacking intact morphology. d Ghost tangles in the EC shown with silver impregnation are surrounded by microglial fragments. e–f iba 1 staining shows detail of fragmented microglial cells in a cluster (center) and in surrounding cells. e Focus series of individual micrographs taken 10–15 μm apart, reassembled into composite image in f. Scale bars 100 μm (a, b, e, f), 50 μm (c, d)

Microglial degeneration can occur independent of age and is evident in cases of young subjects with minimal tau pathology. Double-label immunohistochemistry for microglia (iba1) and tau (AT8) is shown in the hippocampus of two young subjects with no (b) or minimal (d) tau pathology (case nos. 1, 5). a, c Focus series of four individual micrographs taken 10–15 μm apart, and reassembled into composite images in b and d. Note the difference in microglial morphology in the two young subjects, one of which shows minimal tau pathology evident as neuropil threads (arrows in d). Microglia in b show normal ramified appearance but are fragmented in d. Scale bars 20 μm (a, c); 10 μm (b, d)

Single-label (iba1) staining of microglia in two subjects with Down’s syndrome (case nos. 14, 15) is shown in a and b, respectively. Both micrographs reveal a total loss of microglial cell integrity and show presence of microglial cell debris throughout; middle temporal gyrus (a) and entorhinal cortex (b). Scale bar 50 μm
To investigate the temporal relationship between microglial deterioration and neurofibrillary degeneration, we took advantage of the predictability of the spread of tau pathology inherent to Braak staging, namely, that neurodegeneration proceeds predictably from the allocortex (entorhinal and hippocampal areas) to the isocortex [4]. By performing simultaneous analysis of microglial morphology in both the entorhinal cortex and middle temporal gyrus in the same coronal section through the temporal lobe at increasing Braak stages, we were able to determine the extent of microglial dystrophy in regions already showing neurodegeneration as well as in regions that would have developed neurodegeneration if the subject had lived longer. The results, shown in Fig. 5, demonstrate that microglial cells begin their structural deterioration before neurofibrillary pathology sets in. This is particularly evident during Braak stage III where widespread microglial cytorrhexis is coincident with extensive tau pathology in the entorhinal region but also present in the middle temporal gyrus which does not yet show neurodegenerative changes at this stage (Fig. 5g, h). These observations strongly suggest that microglial degeneration precedes the onset of neurofibrillary pathology and therefore support the hypothesis of a causal relationship between the loss of microglial structural integrity and the onset of neurodegeneration.

Microglial fragmentation precedes the spread of tau pathology in the temporal lobe. Double-label immunohistochemistry for microglia (iba1) and tau (AT8) is shown in three subjects with tau pathology increasing from Braak stages 0–III (case nos. 3, 8, and 9). Camera lucida drawings of the actual sections are shown in c, f, and i indicating for orientation the uncus, as well as both sampling areas in the EC and middle temporal gyrus (MTG); areas of tau pathology are shaded in orange. Representative micrographs of the EC (a, d, g) and MTG (b, e, h) reveal microglia (brown) and tau pathology (black) at the different stages. Normal ramified microglia are evident at stage 0 in both EC and MTG in the absence of tau pathology (a, b); mostly fragmented microglia are seen in association with a neurofibrillary tangle and neuropil threads in d, whereas mostly ramified and only a single fragmented cell (arrow) are present in e during stage I; severe microglial fragmentation and loss of discernable cell shape is colocalized with extensive tau pathology in g; microglial processes are fragmented also in h in the absence of neurodegeneration, but cells retain recognizable contours. Scale bar 50 μm (a, b, d, e, g, h)
Given the abundance of literature and seemingly overwhelming evidence supporting a role of Aβ in triggering microglial activation [3, 43], we were compelled to investigate the relationship between Aβ and microglia in situ using the microglial staining procedure employed in this study. In order to do so in a “clean” manner (i.e. without the possibly confounding presence of tau pathology) we selected four subjects who had been diagnosed to have substantial amyloid plaque load (case nos. 16–19; Table 1) but were devoid of tau pathology, and performed double-labeling histochemistry for microglia and Aβ, the latter being visualized using both immunological (antibody 4G8) and silver impregnation techniques (Fig. 6a, b). We found remarkable consistency among these four subjects in that none of them showed any evidence of microglial activation and all were marked by the presence of fully ramified, resting microglia showing even cell spacing without clustering throughout the temporal lobe (Fig. 6a, b). Indeed, it was impressive to observe how non-responsive microglia were to these extensive Aβ deposits which occupied the full extent of cortical gray matter. There were very few instances of dystrophic changes in these cells and the microglial cytorrhexis seen so conspicuously in cases with tau pathology was virtually non-existent. Findings were identical regardless of whether Aβ was demonstrated with 4G8 immunohistochemistry or with the Campbell–Switzer silver method supporting prior observations that immunohistochemical and silver methods for staining Aβ yield near-identical results [5]. As a positive control for purposes of demonstrating microglial activation, we included one subject who was known to have died with sepsis (case no. 6; Table 1). In this subject, microglial activation was rampant and present throughout the full extent of a temporal lobe section (Fig. 6c), yet despite this fulminant neuroinflammation tau pathology was classified as being a minimal Braak stage I, similar to other subjects in this group (case nos. 5,7,8) that showed no neuroinflammation. We interpret these findings to show that (1) Aβ does not cause microglial activation, and (2) microglial activation does not cause or aggravate neurofibrillary degeneration.

Microglial activation is absent in the presence of Aβ but extensive during sepsis. a Double-label immunohistochemistry for microglia (iba1) and Aβ (4G8) reveals fully ramified, non-activated microglia evenly distributed throughout the EC gray matter in the presence of extensive Aβ deposits (case no. 17). Higher magnification reveals full extent of microglial ramification when Aβ is visualized using silver staining in the same subject (b). c Fully activated and hypertrophic microglia are present throughout the EC gray and white matter in a subject with sepsis (case no. 6). Inset Detail of microglial hypertrophy typical for activated microglia. Scale bars 100 μm (a), 50 μm (b), 500 μm (c), 50 μm (inset)
Discussion
The current findings show that the neurodegenerative changes that characterize AD are not accompanied by microglial activation but instead by microglial dystrophy, which likely reflects progressive degeneration of these cells [44]. The significance of these findings is that they provide an explanation as to why anti-inflammatory drugs are ineffective at preventing or diminishing neurodegeneration and dementia. They serve to redirect thinking about AD pathogenesis away from inflammation-induced damage and towards an unexplored area of neuroscience, namely, processes or events that can damage microglial cells. One such process particularly relevant in the context of AD is chronological aging, and it has become clear in the recent years that microglia are subjected to aging-related changes, including telomere shortening [9, 46]. Truncated telomeres in peripheral blood leukocytes also have been identified as a possible marker of increased dementia risk [14]. Since microglia are essential for providing neuroprotection [45], aging-related weakening of microglial neuroprotective function is likely to have detrimental consequences for neurons. We propose here that one such negative consequence is development of neurofibrillary pathology by showing evidence that microglial degeneration likely precedes the onset of tau pathology.
How does one distinguish between resting, activated, and dystrophic microglia? The most reliable way of determination is by a qualitative assessment of cell morphology, which requires robust histochemical demonstration of the cells in situ. The findings shown here are the results of applying an optimal method for staining microglia in human archival material using the iba1 antibody generated by Ito et al. [19]. This method allows the demonstration of all morphological subtypes of microglia, including resting, activated and dystrophic cells which can be distinguished readily based on morphological grounds alone (Fig. 7). Thus, unlike prior studies we have not relied on purportedly specific immunological markers for activated microglia, such as major histocompatibility complex (MHC) antigens, which have long been used to demonstrate ostensible microglial activation (and thus neuroinflammation) in AD and other neurodegenerative diseases [1, 29, 38]. Although an upregulation of MHC antigens occurs on microglia activated after acute injuries, as shown in numerous animal studies, in human brain these antigens (HLA-DR) are widely expressed by non-activated, ramified (resting) microglia, as well as by dystrophic ones [10, 17, 27, 44], and thus anti-MHC staining is a questionable method for the identification of activated microglia [6, 44]. Qualitative morphological assessment also requires viewing microglia at the highest magnification possible for light microscopy and preferably in sections that measure at least 20 μm in thickness, which facilitates viewing in multiples focal planes. Often human brain tissue is examined at low magnification in thin sections of paraffin-embedded tissue, and while this type of analysis may be sufficient for making a diagnosis it does not always afford the level of detail necessary to distinguish between activated and dystrophic, or between resting and dystrophic microglia. The recent analysis by Sasaki et al. [40] claiming a close association between activated microglia and tau pathology underscores this point.

Shown are representative examples of dystrophic (a, b; case no. 9), resting (c, d; case no. 3), activated (e, f; case no. 6) microglial cells at high power. Each set of images shows the same microscopic field in two focal planes using differential interference optics. Dystrophic cells are readily distinguished from both resting and activated cells by their fragmented cytoplasmic processes. Activated cells are distinguished by their hypertrophy. Both resting and activated cells have continuous, non-fragmented processes. All images are from entorhinal cortex. Scale bar 20 μm
The principal feature of microglial dystrophy described in this report is cytoplasmic fragmentation, or cytorrhexis, a process that has not yet been studied in great depth. Cytorrhexis appears to involve the pinching off or budding of cytoplasmic fragments and this phenomenon bears some resemblance to the cytoplasmic changes that occur during apoptosis [20]. However, other prominent features of apoptosis, notably nuclear fragmentation (karyorrhexis), are not apparent during microglial cytorrhexis as the cells show intact nuclei and nucleoli associated with fragmented processes (e.g. Figs. 2, 7). Our previous work describing microglial cytorrhexis in SOD1 transgenic rats also failed to produce evidence in favor of microglial apoptosis [8], and thus we are reluctant to categorize microglial cytorrhexis as either apoptosis or as necrosis at this time. Additional studies are needed to further characterize this seemingly distinct mode of microglial degeneration involving primarily cytoplasmic deterioration. Microglial cytorrhexis occurs in the SOD1G93A rat, a model of amyotrophic lateral sclerosis [8]. Notably, in these animals which die at about 5–7 months of age due to motoneuron loss, the terminal stages of their neurodegenerative condition are marked by prominent microglial cytorrhexis in the spinal cord gray matter where motoneurons are degenerating [8]. Although the mode of motoneuron cell death in SOD1G93A rats remains enigmatic and does not appear to involve neurofibrillary pathology, this model provides an interesting parallel to the current findings in that here too microglial degeneration can be associated with neurodegeneration, thereby supporting our hypothesis that neurodegeneration may be secondary to microglial damage.
Although numerous reports have claimed a role for amyloid peptides in activating microglia and possibly stimulating phagocytosis by microglia (for reviews see [3, 43]), the current findings fail to corroborate the occurrence of either microglial activation or phagocytosis in brains marked by massive Aβ loads. The fact that we did not observe activated microglia in a spectrum of cases ranging from none to severe AD pathology suggests that neither soluble nor insoluble amyloid-beta proteins elicit microglial activation. Since levels of soluble Aβ have been correlated with cognitive impairments [25, 31], our observations do not support the suspected causality between soluble Aβ levels and impaired cognition from a neuroinflammation point of view. However, it is possible that soluble Aβ contributes to microglial degeneration, and this has been shown to occur in vitro under certain conditions [21]. As for neuritic plaques, our findings clearly show that these are accompanied by dystrophic microglia (Fig. 2c), which needs an explanation for the many prior observations reporting activated microglia associated with these lesions [30, 35, 38, 39, 41]. One possibility may be that in prior studies dystrophic microglia were misidentified as activated microglia. This could have been due to that antibodies used in earlier work produced incomplete visualization of microglial cells and also because microglial dystrophy had not yet been recognized. Another possibility may be related to the dynamic structure of Aβ plaques, that is, as these deposits evolve and undergo biochemical changes there may be a point where microglia do become activated and then progress to become dystrophic concurrently with the onset of tau pathology. Yet a third possibility is that the presence of systemic infectious disease may have influenced prior assessments of microglial activation and neuroinflammation in AD brain. Elderly, demented patients often have systemic comorbidities, such as pneumonia and other infections, and it has been shown that infectious disease outside the CNS can profoundly influence microglial activation [23, 27, 36]. Our findings showing pervasive microglial activation in one subject who died with sepsis (case no. 6) seem to confirm this thought, and they underscore the need for more discerning studies of neuroinflammation that take into consideration the absence or presence of peripheral infections.
In summary, the current findings offer an alternative explanation for the involvement of microglia in AD pathogenesis, namely, that a loss of microglial cells contributes to the onset of neurodegeneration. This possibility is appealing because it takes into account old age as a primary factor in the pathogenesis of sporadic AD. According to the microglial dysfunction hypothesis, both microglia and neurons are subject to an aging-related decline in functions and these are exacerbated by genetic and epigenetic factors, including oxidative damage, which may be particularly detrimental to microglia [24]. It remains unclear at this time how exactly damage in microglia is linked to tau pathology in neurons, but this may become a fertile area of investigation in future research. Notwithstanding this as of yet unexplored issue, it is worth noting that in the recent years studies have begun to emerge which provide evidence for microglial abnormalities and degeneration in other neurodegenerative diseases, including amyotrophic lateral sclerosis, Creutzfeldt–Jakob disease, and Huntington’s disease [8, 42, 47], as well as in schizophrenia [48]. Thus, breakdown of the brain’s immune system may be an important factor in the development of neurodegeneration.
References
Aisen PS (1996) Inflammation and Alzheimer disease. Mol Chem Neuropathol 28:83–88
Arvanitakis Z, Grodstein F, Bienias JL et al (2008) Relation of NSAIDs to incident AD, change in cognitive function, and AD pathology. Neurology 70:2219–2225
Boche D, Nicoll JA (2008) The role of the immune system in clearance of Abeta from the brain. Brain Pathol 18:267–278
Braak H, Braak E (1991) Neuropathological staging of Alzheimer-related changes. Acta Neuropathol 82:239–259
Braak H, Braak E (1991) Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol 1:213–216
Croisier E, Moran LB, Dexter DT, Pearce RK, Graeber MB (2005) Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha-synuclein deposition. J Neuroinflamm 2:14
Dickson DW, Crystal HA, Mattiace LA et al (1992) Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging 13:179–189
Fendrick SE, Xue QS, Streit WJ (2007) Formation of multinucleated giant cells and microglial degeneration in rats expressing a mutant Cu/Zn superoxide dismutase gene. J Neuroinflamm 4:9
Flanary BE, Sammons NW, Nguyen C, Walker D, Streit WJ (2007) Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res 10:61–74
Gehrmann J, Banati RB, Kreutzberg GW (1993) Microglia in the immune surveillance of the brain: human microglia constitutively express HLA-DR molecules. J Neuroimmunol 48:189–198
Giulian D, Haverkamp LJ, Yu JH et al (1996) Specific domains of beta-amyloid from Alzheimer plaque elicit neuron killing in human microglia. J Neurosci 16:6021–6037
Graeber MB, Streit WJ (1990) Microglia: immune network in the CNS. Brain Pathol 1:2–5
Grober E, Dickson D, Sliwinski MJ et al (1999) Memory and mental status correlates of modified Braak staging. Neurobiol Aging 20:573–579
Grodstein F, van Oijen M, Irizarry MC et al (2008) Shorter telomeres may mark early risk of dementia: preliminary analysis of 62 participants from the nurses’ health study. PLoS ONE 3:e1590
Hanisch UK, Kettenmann H (2007) Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 10:1387–1394
Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12:383–388
Hayes GM, Woodroofe MN, Cuzner ML (1987) Microglia are the major cell type expressing MHC class II in human white matter. J Neurol Sci 80:25–37
Hickey WF, Kimura H (1988) Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science 239:290–292
Ito D, Imai Y, Ohsawa K et al (1998) Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res 57:1–9
Kerr JF (2002) History of the events leading to the formulation of the apoptosis concept. Toxicology 181–182:471–474
Korotzer AR, Pike CJ, Cotman CW (1993) beta-Amyloid peptides induce degeneration of cultured rat microglia. Brain Res 624:121–125
Kreutzberg GW (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19:312–318
Lemstra AW, Groen in’t Woud JC, Hoozemans JJ et al (2007) Microglia activation in sepsis: a case–control study. J Neuroinflamm 4:4
Lopes KO, Sparks DL, Streit WJ (2008) Microglial dystrophy in the aged and Alzheimer’s disease brain is associated with ferritin immunoreactivity. Glia 56:1048–1060
Lue LF, Kuo YM, Roher AE et al (1999) Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol 155:853–862
Martin BK, Szekely C, Brandt J et al (2008) Cognitive function over time in the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol 65:896–905
Mattiace LA, Davies P, Dickson DW (1990) Detection of HLA-DR on microglia in the human brain is a function of both clinical and technical factors. Am J Pathol 136:1101–1114
McDonald DR, Brunden KR, Landreth GE (1997) Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. J Neurosci 17:2284–2294
McGeer EG, McGeer PL (1998) The importance of inflammatory mechanisms in Alzheimer disease. Exp Gerontol 33:371–378
McGeer PL, Itagaki S, Tago H, McGeer EG (1987) Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett 79:195–200
McLean CA, Cherny RA, Fraser FW et al (1999) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol 46:860–866
Meda L, Cassatella MA, Szendrei GI et al (1995) Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature 374:647–650
Neumann H, Kotter MR, Franklin RJ (2009) Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 132:288–295
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318
Ohgami T, Kitamoto T, Shin RW et al (1991) Increased senile plaques without microglia in Alzheimer’s disease. Acta Neuropathol 81:242–247
Perry VH, Cunningham C, Holmes C (2007) Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol 7:161–167
Riley KP, Snowdon DA, Markesbery WR (2002) Alzheimer’s neurofibrillary pathology and the spectrum of cognitive function: findings from the Nun Study. Ann Neurol 51:567–577
Rogers J, Luber-Narod J, Styren SD, Civin WH (1988) Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer’s disease. Neurobiol Aging 9:339–349
Sasaki A, Yamaguchi H, Ogawa A, Sugihara S, Nakazato Y (1997) Microglial activation in early stages of amyloid beta protein deposition. Acta Neuropathol 94:316–322
Sasaki A, Kawarabayashi T, Murakami T et al (2008) Microglial activation in brain lesions with tau deposits: comparison of human tauopathies and tau transgenic mice TgTau(P301L). Brain Res 1214:159–168
Sheng JG, Mrak RE, Griffin WS (1997) Neuritic plaque evolution in Alzheimer’s disease is accompanied by transition of activated microglia from primed to enlarged to phagocytic forms. Acta Neuropathol 94:1–5
Simmons DA, Casale M, Alcon B et al (2007) Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia 55:1074–1084
Streit WJ (2004) Microglia and Alzheimer’s disease pathogenesis. J Neurosci Res 77:1–8
Streit WJ, Sammons NW, Kuhns AJ, Sparks DL (2004) Dystrophic microglia in the aging human brain. Glia 45:208–212
Streit WJ (2005) Microglia and neuroprotection: implications for Alzheimer’s disease. Brain Res Brain Res Rev 48:234–239
Streit WJ (2006) Microglial senescence: does the brain’s immune system have an expiration date? Trends Neurosci 29:506–510
v Eitzen U, Egensperger R, Kosel S et al (1998) Microglia and the development of spongiform change in Creutzfeldt–Jakob disease. J Neuropathol Exp Neurol 57:246–256
Wierzba-Bobrowicz T, Lewandowska E, Kosno-Kruszewska E et al (2004) Degeneration of microglial cells in frontal and temporal lobes of chronic schizophrenics. Folia Neuropathol 42:157–165
Acknowledgments
This work was supported by the National Institutes of Health Grants AG023665 (WJS) and the German Research Council (Deutsche Forschungsgemeinschaft, DFG) Grant BR 317/17-3 (HB). The authors gratefully acknowledge technical assistance by Mohamed Bouzrou.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Streit, W.J., Braak, H., Xue, QS. et al. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol 118, 475–485 (2009). https://doi.org/10.1007/s00401-009-0556-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-009-0556-6