
Abstract
Hemimegalencephaly (HMEG) is a developmental brain malformation highly associated with epilepsy. Balloon cells (BCs) and cytomegalic neurons (CNs) are frequently observed in HMEG specimens. Cytomegaly in developmental brain malformations may reflect in aberrant activation of the mTOR and β-catenin signaling cascades, known regulators of cell size. We hypothesized that there is aberrant co-expression of phospho-ribosomal S6 (P-S6) protein, a downstream effector of the mTOR cascade, as well as cyclin D1, a downstream effector of the β-catenin pathway, in BCs and cytomegalic neurons in HMEG. We hypothesized that mutations in PTEN (a cause of HMEG associated with Proteus syndrome), TSC1 or TSC2 (tuberous sclerosis complex) genes, which are known to modulate β-catenin and mTOR signaling could cause sporadic HMEG. Expression of cyclin D1, phospho-p70 S6 kinase (P-p70S6K, another mTOR cascade kinase), P-S6, MAP2, NeuN, or GFAP was determined by immunohistochemistry in HMEG brain tissue (n = 7 specimens). Cyclin D1, P-p70S6K, and P-S6 proteins were co-localized in BCs and CNs in the enlarged hemisphere but not in the unaffected hemisphere or in morphologically normal tissue. Cyclin D1 and P-S6 proteins were not detected in GFAP-labeled astrocytes. Sequencing of PTEN, TSC1, and TSC2 genes in cytomegalic cells co-expressing cyclin D1 and P-S6 proteins did not reveal mutations. Selective expression of cyclin D1 and P-S6 in cytomegalic cells in HMEG suggests co-activation of the β-catenin and mTOR cascades. PTEN, TSC1, or TSC2 gene mutations were not detected suggesting that sporadic HMEG is distinct from HMEG associated with Proteus syndrome or tuberous sclerosis complex.
Similar content being viewed by others

Introduction
Hemimegalencephaly (HMEG) is a dramatic malformation of cortical development characterized by marked enlargement and cytoarchitectural abnormalities in one cerebral hemisphere [7, 8, 18]. HMEG is highly associated with early onset intractable epilepsy [7, 8, 18]. Balloon cells (BCs) and cytomegalic neurons (CNs) morphologically similar to those observed in cortical dysplasia (cortical dysplasia with balloon cells, CDBC) or to giant cells (GCs) in tuberous sclerosis complex (TSC) tubers are detected in many, although not in all, HMEG cases [6–8, 18]. The molecular pathogenesis of HMEG remains poorly understood in part because HMEG is typically a sporadic disorder and thus large family pedigrees cannot be assessed. One hypothesis is that HMEG results from a somatic gene mutation occurring in neuroglial progenitor cells during brain development [6].
From a mechanistic prospective, the β-catenin and mTOR (mammalian target of rapamycin) cascades are ideal candidates to study in HMEG because activation of these cellular pathways leads to accelerated cell growth [9, 15, 16]. Previous studies have demonstrated that there is altered β-catenin signaling in CDBC [5]. BCs and CNs in CDBC and hemispheric dysplasia as well as GCs in TSC express phospho-ribosomal S6 (P-S6) protein, a downstream effector of the mTOR cascade [1, 12, 14]. Enhanced expression of activated β-catenin in HMEG is associated with increased levels of cyclin D1, c-myc, and WISP-1 mRNA that are transcriptionally activated by the β-catenin pathway [19]. These results in human tissue are supported experimentally because constitutive activation of β-catenin in transgenic mice leads to megalencephaly [4] and enlarged astrocytes cultured from mice lacking Tsc1 express high levels of P-S6 [19].
We assayed the expression of cyclin D1, phospho-p70 S6 kinase (P-p70S6K), and P-S6 proteins in HMEG as a strategy to determine whether β-catenin and mTOR activation is linked to cytomegaly and hemispheric enlargement. Because HMEG has been reported in TSC patients [3] and in Proteus syndrome patients associated with mutations in the PTEN gene [21], and mTOR is directly modulated by PTEN and the TSC1/TSC2 complex [13, 16], the PTEN, TSC1 and TSC2 genes were sequenced in DNA extracted from HMEG tissue to determine whether aberrant expression of cyclin D1, P-p70S6K, and P-S6 proteins in HMEG results from somatic PTEN or TSC gene mutations.
Methods
Tissue specimens
HMEG specimens (n = 7; 4 males, 3 females; ages 2–24 months) were obtained following epilepsy surgery (n = 5) or at post-mortem examination (n = 2; cause of death was cardiac arrest). One case was associated with linear nevus syndrome, one case with Klippel–Trenaunay–Weber syndrome, and the remaining cases were without any syndromic association. All patients had intractable epilepsy. Histological examination revealed absent cortical lamination, cytomegalic neurons, and BCs throughout the cortex. In two post-mortem cases, the unaffected hemisphere was histologically intact.
Frontal lobe neocortex was obtained from two sets of control patients. First, age-matched normal control cortex was obtained post-mortem (1, 4, 7 months and 1 year, 2 males and 2 females) from individuals without a history of epilepsy. Second, frontal cortex was obtained during epilepsy surgery from patients without cortical malformation (epilepsy control cases; n = 4, mean age 2.7 years, 2 males and 2 females). Studies were approved by the University of Pennsylvania Institutional Review Board.
Immunohistochemistry
Paraffin embedded, 7 μm sections were probed with cyclin D1 (monoclonal rabbit, clone SP4, Lab Vision Corporation, Fremont, CA, USA; 1:100), glial fibrillary acidic protein (GFAP; polyclonal rabbit, DAKO, Glostrup, Denmark; 1:4,000), microtubule-associated protein-2 (MAP2; mouse clone HM2, IgG1; SIGMA, St. Louis, MO, USA; 1:100 and polyclonal rabbit, Chemicon; 1:1,000), neuronal nuclear protein (NeuN; mouse clone MAB377, IgG1; Chemicon, Temecula, CA, USA; 1:1,000), P-S6 protein (Ser235/236; monoclonal rabbit, Cell Signaling Technology, Beverly, MA, USA; 1:100) and P-p70S6K (Thr389; monoclonal mouse; Cell Signaling 1:200), antibodies at 4°C overnight. Single-label immunohistochemistry was visualized using avidin–biotin peroxidase method and 3,3-diaminobenzidine as chromogen.
For double-labeling studies, the cyclin D1 antibody was removed by microwave oven treatment (sodium citrate buffer, 0.01 M, pH 6.0; 650 W for 10 min). Sections were then pre-incubated with 10% goat serum and incubated for 1 h at room temperature with the second primary antibody (GFAP, MAP-2 or P-S6). Following biotinylated secondary antibody, labeling was visualized with the avidin–biotin peroxidase method and liquid permanent red as a chromogen (DAKO). Sections were counterstained with haematoxylin. Digital photomicrographs were generated through contiguous regions of each HMEG sample to define immunolabeling profiles for P-S6 and cyclin D1.
For cyclinD1/NeuN double-labeling studies, sections (after incubation with primary antibodies) were incubated for 2 h with Alexa Fluor® 568 and Alexa Fluor® 488 (anti-rabbit IgG or anti-mouse IgG; 1:200; Molecular probes, Eugene, USA). Sections were then analyzed by means of a laser scanning confocal microscope (Bio-Rad, Hercules, CA, USA; MRC1024) equipped with an argon-ion laser.
Quantitative cell counts
The BCs and CNs were defined using morphometric parameters (maximal cell diameter and process extension) based on NeuN staining for quantitative cell counting analysis applied to immunohistochemically labeled sections. Representative contiguous digital photos were obtained (20× magnification) from each tissue section using image acquisition and analysis software (Spot RT CCD camera, Diagnostic Instruments, Inc. and Phase 3 Imaging System integrated with Image Pro Plus; Media Cybernetics, Silver Spring, MD, USA) [11]. The contiguous images spanned a 1 cm2 region of interest (ROI) in each specimen and were generated under light and phase contrast optics to maximize identification of cell morphology. Prior to final assignment as BCs or CNs by the software, each ROI was visually inspected and any cellular elements erroneously included in the computerized analysis were deleted. Mean maximal diameter (cell diameter at its largest aspect) across the total numbers of morphologically identified BCs and CNs was calculated using Image ProPlus software (expressed in microns by direct calibration with a micrometer). Cytomegaly was operationally defined as a maximal cell soma diameter of greater than 120 μm.
Cell microdissection and PTEN/TSC1/TSC2 gene sequencing in HMEG
For the DNA sequencing studies, single P-S6 immunoreactive BCs (n = 100) visualized under light microscopy were microdissected with CellPixII laser capture system (Arcturus) from each case. We selected BCs for analysis because these cells exhibit the most dramatic cytomegaly in HMEG. Cells were eluted from the dissection caps, and genomic DNA was extracted from the pooled dissected cells (Qiagen DNA extraction kit). Genomic DNA was extracted (Qiagen) from the HMEG specimens or unaffected hemisphere in the two post-mortem cases. Exon-specific flanking PCR primers (forward and reverse) were generated to human TSC1, TSC2 and PTEN genes, and amplicons from all exons were sequenced (ABI Prism). All PCR and sequencing reactions were performed in duplicate and in forward and reverse directions. TSC1/TSC2 sequence results were compared with existing database sequence for TSC1 and TSC2 (http://www.chromium.liacs.nl/lovd/index.php?select_db = TSC1 or db = TSC2) and for PTEN (http://www.ncbi.nlm.nih.gov/Genbank/index.html).
Results
Co-expression of P-S6 and cyclin D1 proteins
The expression of P-S6 and phospho-p70 S6 kinase proteins was nearly absent in normal control cortex, epilepsy control cortex, and the unaffected hemispheres from the two post-mortem HMEG cases (Fig. 1). In contrast, there was robust cytoplasmic P-S6 protein labeling in BCs and CNs present in all seven HMEG cortical specimens (Fig. 1). Cytomegalic neurons exhibited a clear neuronal morphology with apical dendrites and basal axons, whereas BCs exhibited a dysmorphic, ovoid soma, laterally displaced nucleus, and minimal dendritic or axonal extensions. Labeled cells were observed throughout the thickness of the cortex as well as within the heterotopic cells in the subcortical white matter. P-p70S6K expression was also confined to cytomegalic cells in the HMEG cases but was less robust than P-S6 as has been previously reported in TSC [1, 14] (Fig. 2). Because P-S6 is the final endpoint of mTOR cascade activation we used computer-assisted cell counting [see 11] to assess the relationship between P-S6 expression and cytomegaly. Across all ROIs, 94 ± 9% of cells that met morphological criteria for cytomegaly (maximal cell diameter greater than 120 μm) exhibited cytoplasmic P-S6 expression, whereas P-S6 expression in smaller cells (less than 90 μm) was nearly absent. Further subgroup analysis based on morphometric parameters demonstrated that 91 ± 9% of BCs and 81 ± 11% CNs exhibited P-S6 labeling across the ROIs.

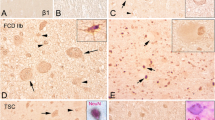
Expression of P-S6 protein in HMEG. a minimal P-S6 expression in control cortex compared with robust expression in HMEG (b scale bar a and b, 500 microns). Panels c–h depict P-S6 expression in cytomegalic neurons (c and inset, d, f, g) and balloon cells (E, F, h, scale bar 60 microns)

Expression of phospho-p70 S6 kinase (P-p70S6K) in HMEG. Note presence of P-p70S6K in cytomegalic cells in low (scale bar, 450 microns) and high (scale bar, 200 microns) power fields
Cyclin D1 expression was not detected in normal control cortex, epilepsy control cortex, or the unaffected hemispheres from the two post-mortem HMEG cases (Fig. 3). Nuclear cyclin D1 labeling was noted in all the HMEG specimens throughout the full thickness of the cortex (Fig. 3). The expression of nuclear cyclin D1 co-localized with P-S6 labeling in BCs and CNs in HMEG across the thickness of the cortex. Many cells that exhibited P-S6 labeling also expressed nuclear cyclin D1, and few cyclin D1-labeled cells were devoid of P-S6 labeling. The expression of nuclear cyclin D1 co-localized with P-S6 labeling in BCs and CNs in HMEG in 87 ± 6% of morphologically identified cytomegalic cells (greater than 120 μm; Fig. 4) but was not detected in the remaining cells less than 100 μm in size.

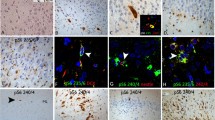
Expression of cyclin D1 in HMEG. a near absence of cyclin D1 expression in control cortex compared with b, robust nuclear expression in HMEG (scale bar a and b, 500 microns). c, d depict higher power views of nuclear cyclin D1 labeling. e, f cyclin D1 expression in neuron confirmed by co-localization of cyclin D1 and NeuN expression (f, and inset). g nuclear cyclin D1 and inset, distinct labeling profile for cyclin D1 (red) and vimentin (green) a marker for immature astrocytes (scale bar e, f, g, 60 microns)

Top co-expression of nuclear cyclin D1 (brown staining) and microtubule associated protein 2 (MAP2, pink staining) in cytomeglic neuron in HMEG. Bottom labeling profiles for cyclin D1, GFAP, and P-S6 protein. a GFAP labeled astrocytes (pink, arrows) do not exhibit cyclin D1 (brown) staining (arrowheads). b–e co-localization of nuclear cyclin D1 (brown) and cytoplasmic P-S6 (pink) staining in cytomegalic neurons (b, d; arrows) and balloon cells (c, e, arrows) HMEG (scale bar 60 microns)
The morphology of many double-labeled cyclin D1 and P-S6 cells was typical of CNs, and in a second set of experiments we show that CNs exhibiting either P-S6 or cyclin D1 labeling co-expressed the neuronal marker proteins, NeuN (Fig. 3) or MAP2 (Fig. 4). In fact, co-labeling of sections with vimentin (a marker for immature astrocytes; Fig. 3) or GFAP (Fig. 4) and either cyclin D1 or P-S6 antibodies demonstrated that vimentin or GFAP-labeled cells exhibiting an astrocytic morphology did not co-express cyclin D1 or P-S6 proteins. In a subpopulation of BCs in all HMEG specimens, vimentin and either P-S6 or cyclin D1 were co-expressed (not shown).
We were unable to make any correlations between clinical phenotype (age, age at seizure onset, duration of seizures, or surgical outcome) and protein marker expression in our sample.
PTEN/TSC1/TSC2 gene sequencing in HMEG
Full exon sequencing of TSC1, TSC2, and PTEN in genomic DNA extracted from HMEG specimens did not reveal mutations. Several previously identified and synonymous PTEN, TSC1 and TSC2 gene sequence polymorphisms were identified in all. In the two post-mortem cases, identical polymorphisms were detected in DNA extracted from the unaffected and enlarged hemispheres suggesting that these sequence alterations were not associated with hemispheric enlargement.
Discussion
We demonstrate that there is aberrant expression of cyclin D1, p70S6K, and P-S6 proteins in HMEG specimens containing BCs and CNs. These proteins were not detected in the morphologically intact hemisphere in two post-mortem HMEG cases suggesting that cyclin D1, P-p70S6K, and P-S6 protein expression are associated with hemispheric enlargement in HMEG. In addition, these results in combination with near absent cyclin D1, P-p70S6K, and P-S6 expression in non-HMEG brain tissue resected from patients with intractable epilepsy argue that expression of these proteins did not reflect the effects of recurrent seizures or anti-epileptic drugs. Co-expression of cyclin D1 and P-S6 proteins overlapped with NeuN and MAP2, but not GFAP, expression suggesting that cyclin D1 and P-S6 protein expression was confined to cells exhibiting neural lineage markers. Sequence analysis did not detect mutations in PTEN, TSC1, or TSC2 in HMEG tissue. However, the enhanced expression of cyclin D1, P-p70S6K, and P-S6 suggests for the first time two cell cascades that may be related to the pathogenesis of HMEG. Since BCs are not a universal finding in HMEG, our results suggest that there may be distinct subtypes of HMEG characterized by the presence or absence of BCs that form by distinct pathogenic mechanisms.
The expression of cyclin D1 and P-S6 was confined to cytomegalic cells providing evidence that there is cell selective activation of these two cascades. In terms of cell lineage, co-expression of cyclin D1 and P-S6 proteins did not overlap with GFAP expression but did co-localize with NeuN expression suggesting that activation of the β-catenin and mTOR cascades was confined to cells exhibiting neural lineage markers. Cyclin D1 expression is transcriptionally activated through β-catenin signaling and is an important marker for cells undergoing mitosis [12, 14]. Increased expression of nuclear cyclin D1 protein in HMEG corroborates our earlier findings of increased cyclin D1 mRNA expression in HMEG and suggests hypothesis that enhanced β-catenin signaling may be related pathogenically to HMEG [20].
The β-catenin and mTOR pathways are linked through TSC1 and TSC2. The TSC1/TSC2 complex promotes β-catenin degradation via association with GSK3 and axin and inhibits Wnt-stimulated gene transcription. Loss of function of TSC1 or TSC2 leads to ribosomal S6 phosphorylation and enhanced β-catenin signaling [15]. Thus, aberrant expression of cyclin D1 and P-S6 in cytomegalic cells in HMEG highlights two pivotal cell growth pathways that may contribute to abnormal cortical development. The fact that more than 90% of cytomegalic cells co-expressed both proteins supports the hypothesis that the aberrant β-catenin and mTOR signaling leads to progressive cellular enlargement in HMEG. Indeed, cells lacking TSC1, TSC2, or PTEN in vitro exhibit progressive cytomegaly [10, 19]. The absence of cyclin D1 and P-S6 in the unaffected hemispheres from two HMEG cases, further demonstrates the potential pathogenic specificity of these pathways in HMEG in relation to enhanced hemispheric and cellular size.
The association between phosphorylation of ribosomal S6 protein and enhanced protein synthesis, increased cell size, and expansive tissue growth has been clearly demonstrated in TSC [1, 17]. Interestingly, mutations in PTEN, which negatively regulates both mTOR and β-catenin signaling [15], have been demonstrated in HMEG associated with Proteus syndrome [19]. Non-synonymous polymorphisms have been reported in PTEN and TSC1 in CDBC [2, 17]; however, no functional PTEN or TSC gene polymorphisms or mutations were detected in our sample of sporadic HMEG cases. These data suggest that sporadic HMEG is genetically distinct from HMEG associated with Proteus syndrome or TSC. A mutation in another candidate gene known to co-regulate both pathways seems plausible.
Enhanced co-expression of cyclin D1 and P-S6 proteins in a subset of HMEG specimens containing BCs and cytomegalic neurons provides a new insight into how HMEG may form during cortical development. These results support the hypothesis that co-activation of the β-catenin and mTOR cascades contributes to cytomegaly and hemispheric enlargement in HMEG.
References
Baybis M, Yu J, Lee A, Golden JA, Weiner H, McKhann G 2nd, Aronica E, Crino PB (2004) mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann Neurol 56(4):478–487
Becker AJ, Urbach H, Scheffler B, Baden T, Normann S, Lahl R, Pannek HW, Tuxhorn I, Elger CE, Schramm J, Wiestler OD, Blumcke I (2002) Focal cortical dysplasia of Taylor’s balloon cell type: mutational analysis of the TSC1 gene indicates a pathogenic relationship to tuberous sclerosis. Ann Neurol 52:29–37
Cartwright MS, McCarthy SC, Roach ES (2005) Hemimegalencephaly and tuberous sclerosis complex. Neurology 64:1634
Chenn A, Walsh CA (2002) Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 297:365–369
Cotter D, Honavar M, Lovestone S, Raymond L, Kerwin R, Anderton B, Everall I (1999) Disturbance of Notch-1 and Wnt signalling proteins in neuroglial balloon cells and abnormal large neurons in focal cortical dysplasia in human cortex. Acta Neuropathol (Berl) 98(5):465–472
Crino PB (2005) Molecular pathogenesis of focal cortical dysplasia and hemimegalencephaly. J Child Neurol 20(4):330–336
Flores-Sarnat L, Sarnat HB, Davila-Gutierrez G, Alvarez A (2003) Hemimegalencephaly: part 2. Neuropathology suggests a disorder of cellular lineage. J Child Neurol 18(11):776–785
Flores-Sarnat L (2002) Hemimegalencephaly: part 1. Genetic, clinical, and imaging aspects. J Child Neurol 17:373–384
Gao X, Pan D (2001) TSC1 and TSC2 tumor suppressors antagonize insulin signaling in cell growth. Genes Dev 15:1383–1392
Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF (2006) Pten regulates neuronal arborization and social interaction in mice. Neuron 50:377–388
Kyin R, Hua Y, Baybis M, Scheithauer B, Kolson D, Uhlmann E, Gutmann D, Crino PB (2001) Differential cellular expression of neurotrophins in cortical tubers of the tuberous sclerosis complex. Am J Pathol 159:1541–1554
Ljungberg MC, Bhattacharjee MB, Lu Y, Armstrong DL, Yoshor D, Swann JW, Sheldon M, D’Arcangelo G (2006) Activation of mammalian target of rapamycin in cytomegalic neurons of human cortical dysplasia. Ann Neurol 60(4):420–429
Mak BC, Takemaru K, Kenerson HL, Moon RT, Yeung RS (2003) The tuberin-hamartin complex negatively regulates Beta-catenin signaling activity. J Biol Chem 278(8):5947–5951
Miyata H, Chiang AC, Vinters HV (2004) Insulin signaling pathways in cortical dysplasia and TSC-tubers: tissue microarray analysis. Ann Neurol 56(4):510–519
Moon RT, Bowerman B, Boutros M, Perrimon N (2002) The promise and perils of Wnt signaling through beta-catenin. Science 296:1644–1666
Persad S, Troussard AA, McPhee TR, Mulholland DJ, Dedhar S (2001) Tumor suppressor PTEN inhibits nuclear accumulation of beta-catenin and T cell/lymphoid enhancer factor 1-mediated transcriptional activation. J Cell Biol 153:1161–1174
Schick V, Majores M, Engels G, Spitoni S, Koch A, Elger CE, Simon M, Knobbe C, Blumcke I, Becker AJ (2006) Activation of Akt independent of PTEN and CTMP tumor-suppressor gene mutations in epilepsy-associated Taylor-type focal cortical dysplasias. Acta Neuropathol (Berl) 112(6):715–725
Tinkle BT, Schorry EK, Franz DN, Crone KR, Saal HM (2005) Epidemiology of hemimegalencephaly: a case series and review. Am J Med Genet A 139(3):204–211
Uhlmann EJ, Li W, Scheidenhelm DK, Gau CL, Tamanoi F, Gutmann DH (2004) Loss of tuberous sclerosis complex 1 (Tsc1) expression results in increased Rheb/S6K pathway signaling important for astrocyte cell size regulation. Glia 47(2):180–188
Yu J, Baybis M, Lee A, McKhann G 2nd, Chugani D, Kupsky WJ, Aronica E, Crino PB (2005) Targeted gene expression analysis in hemimegalencephaly: activation of beta-catenin signaling. Brain Pathol 15(3):179–186
Zhou XP, Marsh DJ, Hampel H, Mulliken JB, Gimm O, Eng C (2000) Germline and germline mosaic PTEN mutations associated with a Proteus-like syndrome of hemihypertrophy, lower limb asymmetry, arteriovenous malformations, and lipomatosis. Hum Mol Genet 9:765–768
Acknowledgment
This work was supported by NS045877 (P.B.C), the National Epilepsy Fund—“Power of the Small”/Hersenstichting Nederland (NEF 02–10; NEF 05–11, E.A. and K.B.) and the Epilepsy Institute of the Netherlands (Heemstede, The Netherlands; E.A.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Aronica, E., Boer, K., Baybis, M. et al. Co-expression of cyclin D1 and phosphorylated ribosomal S6 proteins in hemimegalencephaly. Acta Neuropathol 114, 287–293 (2007). https://doi.org/10.1007/s00401-007-0225-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-007-0225-6