
Abstract
Microbial communities in the active layer play a crucial role in the biogeochemical cycles of Antarctic pristine ecosystems. Here, 16S rRNA gene sequencing was used to investigate bacterial communities in active layer of five different geological sites related to the compositional variation of the geological bedrock, including Neogene volcanic or Cretaceous rocks and or marine sediments areas of distinct elevation. Local variations in the thickness of the active layer (50–80 cm) were observed on the Ulu Peninsula, James Ross Island, and the southwest coast of Vega Island, Antarctica during sampling in 2019. High bacterial diversity was detected in all sampling sites. Significant site effects on bacterial composition with increased Chloroflexota and decreased Flavobacteriaceae were only observed between the highest elevation Johnson Mesa 2 plateau and coastal areas. The overall effect of the depth was reflected by the increased of e.g., Cyanobacteria, Propionibacterium, Staphylococcus in the upper surface and Chloroflexota, Acidobacteriota, Actinomycetota at depths below 30 cm. The huge number of unassigned bacteria indicated a potential source of new bacterial species and their ecological role in this extreme environment. For the first time, we showed that the effect of depth on bacterial composition was more significant than the effect of geological bedrock from these previously unexplored regions.
Similar content being viewed by others
Introduction
Permafrost, a key component of the earth's cryosphere, covers approximately 11% of the world's land area and is typically associated with ground ice (Xue et al. 2019; Obu 2021). The surface layer above permafrost, known as the active layer, thaws seasonally and is considered an important component of terrestrial ecosystems. Active layer monitoring began on a larger scale in the 1990s and more than 100 sites in the Arctic, Antarctica, and several mid-latitude mountain ranges are currently monitored (Hinzman et al. 2003). It plays an important role in cold regions because most ecological, hydrological, biogeochemical and pedogenic (soil-forming) activity takes place within it (Brown et al. 2000). Research on the active layer in Antarctica began in the 1960s in the McMurdo Dry Valleys on Victoria Land, where the largest ice-free areas are located; in other parts of Antarctica, major monitoring of the active layer expanded around the International Polar Year 2007–2009 (Vieira et al. 2010; Hrbáček et al. 2017).
The thickness of the active layer or the existence of permafrost serve as key indicators to describe the terrestrial environment in Antarctica. Due to active layer susceptibility to the effects of climate change, thermal conditions and the thickness of the active layer have been closely monitored in recent decades (Vieira et al. 2010; Bockheim et al. 2013; Hrbáček et al. 2021). The depth of the active layer is spatially variable and depends on location and climate zone, for example, coastal zone sites have a deeper active layer (~ 100 cm), while inland mountain sites have only about 5–10 cm (Adlam et al. 2010; Vieira et al. 2010; Bockheim et al. 2013; Hrbáček et al. 2021).
The extensive deglaciated areas of the Ulu Peninsula, James Ross Island and nearby Vega Island offer the suitable conditions for active layer research. In this unique locality, microbiological research has focused mainly on the description of many novel species comprising phyla Pseudomonadota and Bacteroidota from abiotic sources in last years (Kosina et al. 2016; Sedláček et al. 2017, 2019a, b, 2020, 2021; Švec et al. 2017, 2022; Pantůček et al. 2018; Králová et al. 2019, 2021; Holochová et al. 2020). Only one bacterial composition study was performed between two different soil profiles (60 cm soil profile from Brandy Bay location and 90 cm from Santa Martha Cove) (Meier et al. 2019).
In this study, we have provided a comprehensive view of the variability of microbial composition in soil samples collected from different depths of the active layer from five different geological regions of the Ulu Peninsula, James Ross Island, and Vega Island in Antarctica. The Cape Lachman site, located at the northern end of the Ulu Peninsula, is geologically characterised by the hyaloclastic breccias of the Cape Lachman Formation, which were formed by volcanic activity during the Miocene (Smellie et al. 2008; Mlčoch et al. 2020). The Johnson Mesa area consists of weathered hyaloclastic breccias and subaerial basalts of the Pliocene Johnson Mesa Formation (Smellie et al. 2008; Mlčoch et al. 2020). One sampling location on Johnson Mesa was on the plateau which is located at an elevation of 340 m above sea level, and another was at a lower altitude. The Berry Hill slope area, the foot of the slope below the Berry Hill volcanic mesa, was formed beneath deposits of volcanic material. The geology underlying the Berry Hill slopes profile consists of glacial to marine sedimentary rocks of the Miocene Mendel Formation (Nývlt et al. 2011). The last sampling area was Camp Lamb in the south-western part of Vega Island, which is characterised by marine sedimentary rocks of the Snow Hill Island Formation of Late Cretaceous age and volcanic rocks belonging to the James Ross Volcanic Group of Late Miocene age (Smellie et al. 2008; Roberts et al. 2014).
Materials and methods
Soil samples collected from the James Ross and Vega Islands, Antarctica
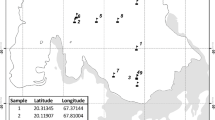
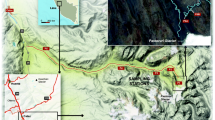
Thirty-one active soil samples were collected from five different locations on the Ulu Peninsula, James Ross Island—Cape Lachman (n = 6), Berry Hill slopes (n = 5), Johnson Mesa 1 (n = 8) and 2 (n = 5), and the southwest coast of Vega Island—Cape Lamb (n = 7), Antarctica, during the austral summer of 2019 (Fig. 1A). The active layer thickness was measured using a probing rod with a diameter of 10 mm. The thickness of the active layer in 2019 varied between sites. Cape Lachman (CL), Berry Hill slopes (BH) and Johnson Mesa 2 (JM2) all had active layer thicknesses of 50 cm, while Vega Island (VI) had 60 cm and, in contrast, Johnson Mesa 1 (JM1) had up to 80 cm. Profiles of all five geological sites were dug until the thaw depth at each location (Fig. 1B) and all active layer samples were then extracted using a small metal shovel inserted horizontally at the desired depth interval in the exposed pit (Fig. 1C). For DNA isolation, a sterile 2 mL tube was filled with approximately 3 g of soil samples.

Locality map and the example of a sampling points. A Map to show positions of the active layer sampling sites on the James Ross and Vega Islands, Antarctica. Five to eight samples were taken from different depths at various geological sites. B All five profiles from the various geological sites on the Ulu Peninsula, James Ross Island and Vega Island, Antarctica were dug until the thaw depth. C All active layer samples were then extracted using a small metal shovel inserted horizontally at the desired depth in the exposed pit
DNA isolation from soil samples, preparation of 16S rRNA gene library and sequencing
A total of 31 soil samples were collected from all five geological sites and different depth intervals. Upon arrival at the laboratory, the samples were frozen at − 20 °C until molecular analyses were performed. The whole ~ 3 g proportion of the sample was homogenized by mixing thoroughly. A 0.2 g soil sample was used for DNA isolation, ensuring that the extracted fraction came from all parts and was representative of the original sample. DNA isolation of all 31 Antarctic soil samples was performed using Pure-Link™ Microbiome DNA Purification Kit (Thermo Fisher Scientific, USA) according to the manufacturer's protocol. The isolated DNA was used as a template in PCR reaction targeting the V4 hypervariable region (515F–806R) of the bacterial 16S rRNA gene (16S Metagenomic sequencing Library Preparation protocol; Illumina, USA). Briefly, the Q5® High-Fidelity 2X Master Mix PCR reactions (New England BioLabs, USA) were carried out in a total volume of 30 μL as follows: 98 °C (30 s), 98 °C (10 s), 55 °C (15 s), 72 °C (25 s), 30 cycles; and 72 °C (2 min). The list of primer pairs is in Table S1. PCR clean-up was performed with SPRIselect beads (Beckman Coulter Genomics, USA). Concentration was measured with Quant-iT dsDNA Assay Kit, High Sensitivity (Thermo Fisher Scientific, USA). Based on the concentration values, samples with different inner tags were equimolarly pooled and used as a template for a second PCR with Nextera XT indexes (Illumina, USA). Next, pools containing different indexes were quantified using the Kapa Library Quantification Kit Illumina Platforms (Roche, USA) and equimolarly pooled. The prepared library was measured with a D1000 Screen tape (Agilent Technologies, USA) and with Kapa Library Quantification Kit Illumina Platforms (Roche, USA). The library was diluted to a final concentration of 8 pM, and 20% of PhiX DNA (Illumina, USA) was added. Sequencing was performed with the MiSeq reagent kit V2 using a MiSeq 2000 instrument according to the manufacturer's instructions (Illumina, USA).
Bioinformatics and statistical analysis
The raw sequence reads were pre-processed by the following bioinformatic pipeline. The first step of the pipeline was demultiplexing of reads in sequencing pools into individual DNA samples. The next step in the pipeline was trimming of low-quality end of each read. Both demultiplexing and length filtering were performed by an in-house tool written in Python 3. Forward and reverse reads were denoised using the DADA2 pipeline version 1.26 for R package (Callahan et al. 2016). Following denoising, the forward and reverse reads were joined using the fastq-join read joining utility (Aronesty 2013). Finally, chimeric sequences were removed from the joined reads using the removeBimera function of the DADA2 R package. Taxonomy was determined using the USEARCH-consensus algorithm from the microbiome analysis toolkit QIIME version 1.9.1 (Caporaso et al. 2010; Edgar 2010). For each input sequence, three closest organisms were found in the Silva version 123 reference database (Quast et al. 2013). Taxonomic names of bacterial phyla obtained from Silva database were corrected according to the publication by Oren and Garrity (2021). Their taxonomies were combined into the final taxonomic assignment using the least common ancestor (LCA) algorithm. Statistical analysis of bacterial compositions was performed at the phylum and genus level. The Shannon diversity index (Ortiz-Burgos 2016) was chosen to estimate the bacterial diversity and uniformity of the microbial community. A Principal Coordinates Analysis (PCoA) plot of the Bray–Curtis distance matrix using the EMPeror visualization tool was used to show differences of bacterial communities between different locations and depths (Vazquez-Baeza et al. 2013). Non-parametric Mann–Whitney U test with Benjamini–Hochberg (BH) adjustment for multiple testing was used to test the differences between soil samples. Results were considered significant at p < 0.05.
Results
A total of 31 Antarctic active layer samples were collected from five different geological locations on the Ulu Peninsula, James Ross Island, and the southwest coast of Vega Island, Antarctica (Fig. 1). Information on the number of reads per sample after quality filtering and chimera removal is in Table S2.
Bacterial diversity in different geological localities and depths of active layer
A high level of bacterial community diversity was observed at all five sampling sites. The Shannon diversity index, which is used to estimate the evenness and richness of microbial populations, averaged approximately 8.3 with no significant differences between sites (Table S2). Lower mean values 7.7 and 7.9 were found in sites CL and VI compared to the most diverse JM2 area (mean Shannon index of 9.6). At sites JM1, JM2 and BH, there was a tendency for diversity to increase with depth, and in the deepest layers of these localities (near the permafrost), Shannon index values were the highest and exceeded 10.
Bacterial compositional changes according to the geological location
At the phylum level, Actinomycetota, Pseudomonadota, Bacillota, Bacteroidota, and Chloroflexota were the most abundant in all geological sites (Fig. 2; Table S3). Statistically significant differences were described in the JM2 area, where a higher abundance of Chloroflexota was found compared to CL (p = 0.04) and VI (p = 0.02). An increased abundance of Flavobacteriaceae was detected in the coastal Cretaceous VI area compared to area JM2 (p = 0.03).

Localization and metagenomic analysis (at the phylum level) of 31 soil samples isolated from different depths of active layer from five various geological sites on the Ulu Peninsula, James Ross Island and Vega Island, Antarctica. *Other—bacteria with an average value in all samples below 1%
At the genus level, Propionibacterium, Corynebacterium (both, with the highest level detected at the CL locality), and Staphylococcus dominated at all sites (Fig. 3A; Table S3). From the results previously described using the Shannon index, it was already evident that bacterial richness would also be very high. Many different genera were detected at all sampling sites, with a total abundance of approximately 1–2%, except for the two or so dominant genera, which only accounted for approximately ten percent of the total abundance. Also, many genera corresponding to uncultivated and unassigned taxa were found.

Bacterial communities at different depths of the active layer. A Bacterial abundance at the genus level. *The bacterial profile of the last three deepest layers of JM2 was like the last two lower layers of JM1. **Other—the 30 most abundant bacterial genera are visualized, for other see Table S3. B A Principal Coordinates Analysis (PCoA) plot of the Bray–Curtis distance matrix using the differences of bacterial communities between different locations and depths of active layer and clustering
Relationship between the bacterial composition and soil depth
According to the PCoA plot, two main clusters of samples were identified (Fig. 3B). The first cluster mainly included samples from the deepest layers of JM1 and JM2 along with some samples from other sites also from deeper layers (30 cm or deeper). The second cluster grouped samples from the uppermost layers of JM1 and JM2 together with a variety of primarily uppermost samples from other sites (BH, VI and CL). The bacterial profile of the last three deepest layers of JM2 was like that of the last two lower layers of JM1 (Fig. 3A).
The results from the PCoA plot showed the potential effect of the soil depth on the microbial communities. Overall, at all sites, the abundance of Chloroflexota increased significantly at 30–40 cm depth (p = 0.04). Acidobacteriota abundance gradually increased from the active layer surface, and the highest values were found at a depth of approximately 40 cm compared to the upper (p = 0.02) and deeper layers (p = 0.02). Cyanobacteria were increased in the 0–10 cm compared to 20–50 cm (p = 0.03).
At the genus level, Propionibacterium (p = 0.003), Staphylococcus (p = 0.01), Corynebacterium (p = 0.003), Streptococcus (p = 0.004), Enterobacteriaceae unassigned (p = 0.02) were significantly increased in the upper 0–30 cm layer compared to the lower parts. While Acidimicrobiales uncultured (p = 0.04), Chloroflexota KD4-96 uncultured (p = 0.04), Thermomicrobiota JG30-KF-CM45 (p = 0.02), Chloroflexota Gitt-GS-136 unassigned (p = 0.02), Gemmatimonadaceae uncultured (p = 0.02), Actinomycetota MB-A2-108 uncultured (p = 0.02), and Thermophilia-Solirubrobacterales 480-2 uncultured (p = 0.02) abundances were increased in the layers below 30 cm compared to upper layers.
Discussion
The Ulu Peninsula, located in the northern part of James Ross Island, is the most extensive deglaciated area in the Antarctic Peninsula (Hrbáček et al. 2017). While deglaciation of the Ulu Peninsula, James Ross Island, began as early as ~ 13 ka bp (Nývlt et al. 2014), the area on the Vega Island, where another sampling site was located, deglaciate between 8 and 6 ka bp (Píšková et al. 2019). Since 2010, the number of studies of active layer dynamics in the Antarctic region has increased. Most of these studies have focused on investigating the thermal conditions of the active layer through drilling in soil or sediments, as well as its relation to climate (Vieira et al. 2010; Bockheim et al. 2013; Hrbáček et al. 2021).
The active layer of soil above the permafrost thaws during the austral summer months (Amesbury et al. 2017). This first study which focuses on the bacterial composition of active layer from soil samples collected in five geologically distinct locations on the Ulu Peninsula and the southwest coast of Vega Island can help to understand how the active layer will respond to climate change, and what the implications of those changes might be for the broader ecosystem. Microbial diversity in all study areas was very high compared to previously studied sites at Brandy Bay and Santa Marta Cove with Shannon index approximately 4.2 (Meier et al. 2019). The highest diversity was described in the deepest layers of areas Johnson Mesa 1, Johnson Mesa 2, and Berry Hill slopes. Some studies suggest that dead intact cells and extracellular genomic DNA may increase the diversity of soil microorganisms (Carini et al. 2016). In this deepest part of the active layer above the permafrost, seasonal thawing probably leads to the accumulation of various bacteria or DNA on this surface. The higher diversity values from the deepest layers may be related to the fact that this is probably a historically active layer, as in the previous years, 2006–2015, the active layer was approximately 5–15 cm shallower (Hrbáček et al. 2017).
Differences in the active layer thickness were found between the different geological sites. The site in Berry Hill slopes area has active layer thickness around 90 cm (Hrbáček et al. 2017), but excavation of the soil profile was problematic as it could only be dug to a maximum depth of 50 cm due to subsequent water flooding. Johnson Mesa 1 is located on the north-aspect sunny slope and consists of volcanic material that effectively traps heat. This explains that the deepest layer was sampled in this site. The Cape Lachman area is also composed of volcanic material, but the sampling site is south-facing and was probably cooler compared to Johnson Mesa 1 site. Johnson Mesa 2 is located on a plateau; it is the highest elevation area and therefore likely has a thinner active layer than Johnson Mesa 1 site. The Cretaceous coastal area of Vega Island, like the coastal area of Cape Lachman, has a similar thickness of the active layer.
Except for Johnson Mesa 2, which differed from Cape Lachman and Vega Island in having significantly elevated Chloroflexota and from Vega Island in having reduced Flavobacteriaceae abundance, no other statistically significant differences were described between the areas. The Johnson Mesa 2 area localized on the plateau was likely influenced by the higher elevation compared to Johnson Mesa 1, localized 30 m lower. Also, the thickness of active layer was smaller in the plateau area and the bacterial composition of the last two samples from the 70 to 80 cm of Johnson Mesa 1 was like the composition below the 30 cm depth in the higher Johnson Mesa 2 plateau area. Of all the areas studied, Johnson Mesa 2 and Vega Island differed the most from each other. Cretaceous coastal Vega Island area probably varied from the Johnson Mesa 2 mesa plateau area in the salinity and chloride concentrations, and this is possibly responsible for the increased Flavobacteriaceae in Vega Island, which represent a group of adapted psychro- and halotolerant bacteria (Bajerski and Wagner 2013; Meier et al. 2019).
All active layer soils were dominated by Actinomycetota, Pseudomonadota, Bacillota, Bacteroidota, and Chloroflexota, which is in accordance with previously published results in other Antarctic habitats—different soils (vegetated sites, carbon-depleted soils, mineral soils, cryosols), glacier forefields and rocks, where these bacterial phyla also predominated in varying order and abundance (Ganzert et al. 2011; Bajerski and Wagner 2013; Meier et al. 2019; Garrido-Benavent et al. 2020). The depth of the active layer, as opposed to the influence of the geological location, emerged as a key factor determining the distribution of individual bacteria. Higher abundances of Cyanobacteria, Propionibacterium, Staphylococcus, Corynebacterium, Streptococcus, and Enterobacteriaceae were observed in the higher layers, while Chloroflexota, Acidobacteriota, Acidimicrobiales, Thermomicrobiota, Gemmatimonadaceae, Actinomycetota, and “Thermophilia” in the layers below 30 cm. It has been previously published that Cyanobacteria dominate the surface layers, while the deeper layers are inhabited by phototrophic bacteria from the phyla Chlorobiota and Chloroflexota (Jungblut and Vincent 2017). Surprisingly, we found a high abundance of mainly human skin-related bacteria, such as Propionibacterium, Staphylococcus, Corynebacterium, and Streptococcus in the upper layers. Although these bacteria represent several known contaminants of microbiota studies (Salter et al. 2014), our negative sequencing controls did not contain these bacteria and the confirmation of the finding of many of these genera in the avian microbiome (Hird et al. 2015) point to birds as a possible source of soil microbiome. Pearce et al. (2009) also described the presence of these microorganisms in the air samples over Antarctica. Air together with birds could probably represent a significant source of these bacteria in the upper parts of the soil active layer which can also into the lower layers with the assistance of water. Similarly like in our study, Meier et al. (2019) also detected higher values in Gemmatimonadaceae in the deeper parts of Brandy Bay soil and suggested that there might be a connection with the water availability. The huge number of uncultured and unassigned bacteria indicated a significant source of new bacterial species and their potential, e.g., as a promising source of new antimicrobial compounds.
In summary, our findings describe that the active layer of the deglaciated areas of the Ulu Peninsula and Vega Island contain a huge and diverse bacterial community. For the first time, we compared soil samples from different depths of active layer from these previously unexplored areas and showed that the effect of depth on bacterial composition was more significant than the effect of geological sites.
Data availability
The data underlying this article are available in the article and in its Online Supplementary Material. The BioProject accession number associated with described metagenomic data are PRJNA961061. BioSample accession numbers are listed in Table S2.
References
Adlam L, Balks M, Seybold C, Campbell D (2010) Temporal and spatial variation in active layer depth in the McMurdo Sound Region, Antarctica. Antarct Sci 22:45–52. https://doi.org/10.1017/S0954102009990460
Amesbury MJ, Roland TP, Royles J, Hodson DA, Convey P, Griffiths H, Charman DJ (2017) Widespread biological response to rapid warming on the Antarctic Peninsula. Curr Biol 27:1616–1622. https://doi.org/10.1016/j.cub.2017.04.034
Aronesty E (2013) Comparison of sequencing utility programs. Open Bioinform J 7:1–8. https://doi.org/10.2174/1875036201307010001
Bajerski F, Wagner D (2013) Bacterial succession in Antarctic soils of two glacier forefields on Larsemann Hills, East Antarctica. FEMS Microbiol Ecol 85:128–142. https://doi.org/10.1111/1574-6941.12105
Bockheim J, Vieira G, Ramos M, López-Martínez J, Serrano E, Guglielmin M, Wilhelm K, Nieuwendam A (2013) Climate warming and permafrost dynamics in the Antarctic Peninsula region. Glob Planet Change 100:215–223. https://doi.org/10.1016/j.gloplacha.2012.10.018
Brown J, Hinkel KM, Nelson FE (2000) The Circumpolar Active Layer Monitoring (CALM) program: research designs and initial results. Polar Geogr 24:166–258. https://doi.org/10.1080/10889370009377698
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. https://doi.org/10.1038/nmeth.3869
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. https://doi.org/10.1038/nmeth.f.303
Carini P, Marsden PJ, Leff JW, Morgan EE, Strickland MS, Fierer N (2016) Relic DNA is abundant in soil and obscures estimates of soil microbial diversity. Nat Microbiol 2:16242. https://doi.org/10.1038/nmicrobiol.2016.242
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. https://doi.org/10.1093/bioinformatics/btq461
Ganzert L, Lipski A, Hubberten H-W, Wagner D (2011) The impact of different soil parameters on the community structure of dominant bacteria from nine different soils located on Livingston Island, South Shetland Archipelago, Antarctica. FEMS Microbiol Ecol 76:476–491. https://doi.org/10.1111/j.1574-6941.2011.01068.x
Garrido-Benavent I, Pérez-Ortega S, Durán J, Ascaso C, Pointing SB, Rodríguez-Cielos R, Navarro F, de Los RA (2020) Differential colonization and succession of microbial communities in rock and soil substrates on a maritime Antarctic glacier forefield. Front Microbiol 11:126. https://doi.org/10.3389/fmicb.2020.00126
Hinzman LD, Fukuda M, Sandberg DV, Chapin FS III, Dash D (2003) FROSTFIRE: an experimental approach to predicting the climate feedbacks from the changing boreal fire regime. J Geophys Res Atmos 108:8153. https://doi.org/10.1029/2001JD000415
Hird SM, Sánchez C, Carstens BC, Brumfield RT (2015) Comparative gut microbiota of 59 Neotropical bird species. Front Microbiol 6:1403. https://doi.org/10.3389/fmicb.2015.01403
Holochová P, Mašlaňová I, Sedláček I, Švec P, Králová S, Kovařovic V, Busse HJ, Staňková E, Barták M, Pantůček R (2020) Description of Massilia rubra sp. nov., Massilia aquatica sp. nov., Massilia mucilaginosa sp. nov., Massilia frigida sp. nov., and one Massilia genomospecies isolated from Antarctic streams, lakes and regoliths. Syst Appl Microbiol 43:126112. https://doi.org/10.1016/j.syapm.2020.126112
Hrbáček F, Nývlt D, Láska K (2017) Active layer thermal dynamics at two lithologically different sites on James Ross Island, Eastern Antarctic Peninsula. CATENA 149:592–602. https://doi.org/10.1016/j.catena.2016.06.02
Hrbáček F, Engel Z, Kňažková M, Smolíková J (2021) Effect of summer snow cover on the active layer thermal regime and thickness on CALM-S JGM site, James Ross Island, eastern Antarctic Peninsula. CATENA 207:105608. https://doi.org/10.1016/j.catena.2021.105608
Jungblut AD, Vincent WF (2017) Cyanobacteria in polar and alpine ecosystems. In: Margesin R (ed) Psychrophiles: from biodiversity to biotechnology. Springer, Cham. https://doi.org/10.1007/978-3-319-57057-0_9
Kosina M, Švec P, Černohlávková J, Barták M, Snopková K, De Vos P, Sedláček I (2016) Description of Pseudomonas gregormendelii sp. nov., a novel psychrotrophic bacterium from James Ross Island, Antarctica. Curr Microbiol 73:84–90. https://doi.org/10.1007/s00284-016-1029-5
Králová S, Busse HJ, Švec P, Mašlaňová I, Staňková E, Barták M, Sedláček I (2019) Flavobacterium circumlabens sp. nov. and Flavobacterium supremum sp. nov., two psychrotrophic species isolated from Antarctic environmental samples. Syst Appl Microbiol 42:291–301. https://doi.org/10.1016/j.syapm.2018.12.005
Králová S, Busse HJ, Švec P, Mašlaňová I, Staňková E, Barták M, Sedláček I (2021) Flavobacterium flabelliforme sp. nov. and Flavobacterium geliluteum sp. nov., two multidrug-resistant psychrotrophic species isolated from Antarctica. Front Microbiol 12:729977. https://doi.org/10.3389/fmicb.2021.729977
Meier LA, Krauze P, Prater I, Horn F, Schaefer CEGR, Scholten T, Wagner D, Mueller CW, Kühn P (2019) Pedogenic and microbial interrelation in initial soils under semiarid climate on James Ross Island, Antarctic Peninsula region. Biogeosciences 16:2481–2499. https://doi.org/10.5194/bg-16-2481-2019
Mlčoch B, Nývlt D, Mixa P (2020) Geological map of James Ross Island—Northern Part 1: 25000. Czech Geological Survey, Praha
Nývlt D, Košler J, Mlčoch B, Mixa P, Lisá L, Bubík M, Hendriks BWH (2011) The Mendel formation: evidence for Late Miocene climatic cyclicity at the northern tip of the Antarctic Peninsula. Palaeogeogr Palaeoclimatol Palaeoecol 299:363–384. https://doi.org/10.1016/j.palaeo.2010.11.017
Nývlt D, Braucher R, Engel Z, Mlčoch B, ASTRE Team (2014) Timing of the Northern Prince Gustav Ice Stream retreat and the deglaciation of northern James Ross Island, Antarctic Peninsula during the last glacial–interglacial transition. Quat Res 82:441–449. https://doi.org/10.1016/j.yqres.2014.05.003
Obu J (2021) How much of the Earth's surface is underlain by permafrost? J Geophys Res-Earth 126:e2021JF006123. https://doi.org/10.1029/2021JF006123
Oren A, Garrity GM (2021) Valid publication of the names of forty-two phyla of prokaryotes. Int J Syst Evol Microbiol 71:005056. https://doi.org/10.1099/ijsem.0.005056
Ortiz-Burgos S (2016) Shannon–Weaver diversity index. In: Kennish MJ (ed) Encyclopedia of estuaries. Encyclopedia of earth sciences series. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-8801-4_233
Pantůček R, Sedláček I, Indráková A, Vrbovská V, Mašlaňová I, Kovařovic V, Švec P, Králová S, Krištofová L, Kekláková J, Petráš P, Doškař J (2018) Staphylococcus edaphicus sp. nov., isolated in Antarctica, harbors the mecC gene and genomic islands with a suspected role in adaptation to extreme environments. Appl Environ Microbiol 84:e01746-e1817. https://doi.org/10.1128/AEM.01746-17
Pearce DA, Bridge PD, Hughes KA, Sattler B, Psenner R, Russell NJ (2009) Microorganisms in the atmosphere over Antarctica. FEMS Microbiol Ecol 69:143–157. https://doi.org/10.1111/j.1574-6941.2009.00706.x
Píšková A, Roman M, Bulinova M, Pokorný M, Sanderson DCW, Cresswell A, Juan ML, Coria SH, Nedbalová L, Lami A, Musazzi S, de Vijver BV, Nývlt D, Kopalová K (2019) Late-Holocene palaeoenvironmental changes at Lake Esmeralda (Vega Island, Antarctic Peninsula) based on a multi-proxy analysis of laminated lake sediment. Holocene 29:095968361983803. https://doi.org/10.1177/0959683619838033
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590-596. https://doi.org/10.1093/nar/gks1219
Roberts EM, Lamanna MC, Clarke JA, Meng J, Gorsack E, Sertich JJW, O’Connor PM, Claeson KM, MacPhee Ross DE (2014) Stratigraphy and vertebrate paleoecology of Upper Cretaceous? Lowest Paleogene strata on Vega Island, Antarctica. Palaeogeogr Palaeoclimatol Palaeoecol 402:55–72. https://doi.org/10.1016/j.palaeo.2014.03.005
Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, Turner P, Parkhill J, Loman NJ, Walker AW (2014) Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol 12:87. https://doi.org/10.1186/s12915-014-0087-z
Sedláček I, Králová S, Kýrová K, Mašlaňová I, Busse HJ, Staňková E, Vrbovská V, Němec M, Barták M, Holochová P, Švec P, Pantůček R (2017) Red–pink pigmented Hymenobacter coccineus sp. nov., Hymenobacter lapidarius sp. nov. and Hymenobacter glacialis sp. nov., isolated from rocks in Antarctica. Int J Syst Evol Microbiol 67:1975–1983. https://doi.org/10.1099/ijsem.0.001898
Sedláček I, Pantůček R, Holochová P, Králová S, Staňková E, Vrbovská V, Šedo O, Švec P, Busse HJ (2019a) Hymenobacter humicola sp. nov., isolated from soils in Antarctica. Int J Syst Evol Microbiol 69:2755–2761. https://doi.org/10.1099/ijsem.0.003555
Sedláček I, Pantůček R, Králová S, Mašlaňová I, Holochová P, Staňková E, Vrbovská V, Švec P, Busse HJ (2019b) Hymenobacter amundsenii sp. nov. resistant to ultraviolet radiation, isolated from regoliths in Antarctica. Syst Appl Microbiol 42:284–290. https://doi.org/10.1016/j.syapm.2018.12.004
Sedláček I, Pantůček R, Zeman M, Holochová P, Šedo O, Staňková E, Švec P, Králová S, Vídeňská P, Micenková L, Urvashi KS, Lal R (2020) Hymenobacter terrestris sp. nov. and Hymenobacter lapidiphilus sp. nov., isolated from regoliths in Antarctica. Int J Syst Evol Microbiol 70:6364–6372. https://doi.org/10.1099/ijsem.0.004540
Sedláček I, Holochová P, Sobotka R, Busse HJ, Švec P, Králová S, Šedo O, Pilný J, Staňková E, Koublová V, Sedlář K (2021) Classification of a violacein-producing psychrophilic group of isolates associated with freshwater in Antarctica and description of Rugamonas violacea sp. nov. Microbiol Spectr 9:e00452-21. https://doi.org/10.1128/Spectrum.00452-21
Smellie JL, Johnson JS, McIntosh WC, Esser R, Gudmundsson MT, Hambrey MJ, van Wyk de Vries B (2008) Six million years of glacial history recorded in volcanic lithofacies of the James Ross Island Volcanic Group, Antarctic Peninsula. Palaeogeogr Palaeoclimatol Palaeoecol 260:122–148. https://doi.org/10.1016/j.palaeo.2007.08.011
Švec P, Králová S, Busse HJ, Kleinhagauer T, Pantůček R, Mašlaňová I, Cnockaert M, Vandamme P, Staňková E, Gelbíčová T, Holochová P, Barták M, Kýrová K, Sedláček I (2017) Pedobacter jamesrossensis sp. nov., Pedobacter lithocola sp. nov., Pedobacter mendelii sp. nov., and Pedobacter petrophilus sp. nov., isolated from Antarctic environment. Int J Syst Evol Microbiol 67:1499–1507. https://doi.org/10.1099/ijsem.0.001749
Švec P, Králová S, Staňková E, Holochová P, Sedlář K, Koudelková S, Krsek D, Grzesiak J, Šedo O, Váczi P, Urvashi, Gupta V, Sood U, Lal R, Korpole S, Sedláček I (2022) Pedobacter fastidiosus sp. nov., isolated from glacial habitats of maritime Antarctica. Int J Syst Evol Microbiol 72:005309. https://doi.org/10.1099/ijsem.0.005309
Vazquez-Baeza Y, Pirrung M, Gonzalez A, Knight R (2013) EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience 2:16. https://doi.org/10.1186/2047-217X-2-16
Vieira G, Bockheim J, Guglielmin M, Balks M, Abramov AA, Boelhouwers J, Cannone N, Ganzert L, Gilichinsky DA, Goryachkin S, López-Martínez J, Meiklejohn I, Raffi R, Ramos M, Schaefer C, Serrano E, Simas F, Sletten R, Wagner D (2010) Thermal state of permafrost and active-layer monitoring in the antarctic: advances during the international polar year 2007–2009. Permafr Periglac Process 21:182–197. https://doi.org/10.1002/ppp.685
Xue Y, Jonassen I, Øvreås L, Taş N (2019) Bacterial and archaeal metagenome-assembled genome sequences from svalbard permafrost. Microbiol Resour Announc 8:e00516-19. https://doi.org/10.1128/MRA.00516-19
Acknowledgements
The authors would like to thank the Czech Antarctic Research Programme and its crew for providing support and to Iva Buriánková and Matěj Roman for their help with soil sampling (IB—Vega Island and Cape Lachman and MR—collaboration in the Johnson Mesa area).
Funding
Open access publishing supported by the National Technical Library in Prague. This work was supported by the Projects CzechPolar2 RI by the Ministry of Education, Youth and Sports of the Czech Republic (LM2015078) and National Institute of Virology and Bacteriology (Programme EXCELES, ID Project LX22NPO5103)—Funded by the European Union—Next Generation EU. Authors thank the Research Infrastructure RECETOX RI (LM2023069) financed by the Ministry of Education, Youth and Sports for supportive background. This study was also partly funded by the Czech Collection of Microorganisms. The work of FH was supported by Czech Science Foundation Project (GM22-28659M).
Author information
Authors and Affiliations
Contributions
IS, FH, PŠ and DN designed the research and determined sample collection. IM and LM processed the samples. RP, LM and IM interpreted the data. LM and IM prepared the manuscript. All authors read, reviewed, and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
None declared.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Micenková, L., Sedláček, I., Hrbáček, F. et al. Effect of soil depth on the structure of bacterial composition in the active layer at five geologically distinct sites on James Ross and Vega Islands in Antarctica. Polar Biol 47, 295–303 (2024). https://doi.org/10.1007/s00300-024-03230-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00300-024-03230-3