
Abstract
Most infectious agents, such as viruses, bacteria and parasites, can trigger autoimmunity via different mechanisms. The development of an autoimmune disorder after infection tends to occur in genetically susceptible individuals. Some parameters, such as genetic predisposition, feature of the infectious agent and sometimes protective effect of the infections, have a significant role in this process. These parameters and various pathogens that could lead to enhancement or exacerbation of autoimmune disease were examined in this review. Recent studies were reviewed from a microbiological perspective.
Similar content being viewed by others


Introduction
Autoimmune disease is a group of disorders in which the primary cause is the inflammatory reaction caused by the body’s own immune system attacking tissues.
Autoimmune diseases are the third most common category of disease in the United States after cancer and heart disease; they affect approximately 5–8 % of the population or 14–22 million persons [1]. Epidemiologic studies on the autoimmune disease indicated that prevalence of the diseases on the world showed regional differences. Autoimmune diseases can affect every site in the body, including the endocrine system, connective tissue, gastrointestinal tract, heart, skin and kidneys. At least 15 diseases are known to be the direct result of an autoimmune response, while circumstantial evidence implicates >80 conditions with autoimmunity [2].
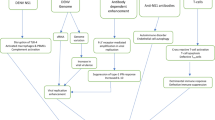
Comprehending the autoimmune diseases is obstructed by the fact that some level of autoimmunity, in the form of naturally occurring autoantibodies and self-reactive T and B cells, is present in all normal persons [3]. An autoimmune response occurs in most persons, but clinically relevant autoimmune disease develops only in susceptible persons. Can “molecular mimicry,” be the reason? Can infections trigger autoimmune disease? How can infections induce autoimmune disease?
Autoimmune diseases sometimes occur shortly after suffering from infectious diseases. To explain the connection between infection and autoimmunity, both antigen-specific and antigen-nonspecific mechanisms have been suggested [4]. For instance, a number of infectious agents have been proposed as possible triggering factors in systemic sclerosis. Four pathogenic hypotheses have been proposed: molecular mimicry, endothelial cell damage, super-antigens, and microchimerism [5].
Molecular mimicry
Molecular mimicry is among the most popular theories about how virus may induce autoimmunity [6]. Structural molecular mimicry occurs when a viral peptide has an amino acid sequence similar or identical to an amino acid sequence of a self peptide, resulting in cross-reactive T cell and B cell responses. A potentially autoreactive T cell, possessing T cell receptors that recognize both a foreign (viral) peptide and a self-peptide, is activated by a virus-derived peptide. Thus, in addition to mediating an antiviral response, the T cell is also capable of mediating self-directed responses [7]. The antigen-presenting cells (macrophages, dendritic cells, microglia) as well as the capability of the mimic peptide, being processed from the native pathogen protein, are two key factors that play important role in the molecular mimicry mechanism during the induction of autoimmunity. In addition, the nature of the innate immune response to the pathogen that determines the immunopathologic potential of the induced cross-reactive T cells, the site of the primary infection in the host and the ability of the pathogen to persist, and the potential requirement for multiple infections with the same or different pathogens, are all considered as contributing factors determining the mechanism of molecular mimicry [8].
There has been a long-standing association between infectious organisms and human autoimmunity. Two major theories have been proposed to account for the correlation between infection and autoimmune disease. The antigen-specific theory is explained by the concept of molecular mimicry in which microbial antigens and self-antigens share structural similarities. The antigen-nonspecific theory assigns a primary role to “bystander activation”, in which the immune system can be activated by either the abnormal release of endogenous proteins as a consequence of microbe-induced cell death, or by antigen-presenting cells activated by the microbial stimulus to more effectively present self-antigen. It is also possible that direct engagement of toll-like receptors (TLRs) by inappropriately released or modified self-determinants may be sufficient to trigger an autoimmune response in the absence of infection [9]. TLRs are a major class of germ-line encoded receptors that activate the immune system in response to a variety of pathogen-associated molecular patterns (PAMPs). PAMPs contain conserved molecular motifs that are generally found in microorganisms, but not in their eukaryotic hosts. TLRs have a broad, heterogeneous tissue expression pattern, and their ligands can mediate effects on a various cell types. The TLR can bind a range of microbial components from bacteria, fungi, and viruses. In addition to mediating protective immunity against pathogens, TLR stimulation may potentially contribute to autoimmune responses. Exogenous, microbial nucleic acids can exacerbate SLE pathology presumably through TLR stimulation. TLRs distinguishing between nucleic acids of microbial and mammalian origin are imperfect so that endogenous, non-microbial nucleic acids can also potentially act as TLR ligands under certain conditions. As TLRs recognize nucleic acids and modulate B cell effector functions, it might be predicted that they would have a role in the activation of autoimmune B cells in SLE. The potential involvement of TLRs is not limited to the direct activation of B cells. Growing evidence suggests that TLR recognition of nucleic acids in SLE immune complexes could also contribute to activation of the innate immune system [10, 11].
Endothelial cell damage
In most adult organisms, endothelial cells are quiescent with turnover rates estimated to be in the order of years, with the exception of the reproductive cycle in fertile females and in wound healing or tissue regeneration [12]. However, endothelial cell proliferation and new vessel formation are characteristics of several diseases such as cancer and macular degeneration, while, on the other hand, endothelial cell death is also a typical feature of diseases such as atherosclerosis, allograft vasculopathy, heart failure, diabetic retinopathy and systemic sclerosis (SSc) [13]. In addition, there is ample evidence that endothelial dysfunction occurs in rheumatoid arthritis (RA) [14]. Intracellular adhesion molecule, E-selectin and L-selectin are upregulated in rheumatoid arthritis and correlate with inflammatory markers [15]. Not only do RA patients express adhesion molecules in synovium, which enables leukocytes to migrate there and cause inflammation, circulating concentrations of several cell adhesion molecules are increased, a sign of endothelial activation [16]. In SSc pathogenesis, chronic inflammation plays a role in endothelial cell aging and damage. It is likely that prolonged endothelial cell perturbation and activation may lead to dysfunction and irreversible loss of integrity, with cell detachment and persistent tissue injury endothelial cell damage with apoptosis resulting in the loss of capillaries is considered as one of the earliest changes in the pathogenesis of SSc [17]. Other mechanism is oxidative stress. Oxidative stress is associated with endothelial cell aging, due to a progressive reduction of the endogenous free radical scavengers over time. Chronic exposure of endothelial cells to radical oxygen species induces morphological changes and impairment of cell–cell adhesion. Oxidative stress also increases vascular endothelial permeability, which is coupled with alterations in endothelial cell signal transduction [13].
Superantigens
Superantigens are proteins produced by a variety of microorganisms, especially bacteria or mycoplasma, or virus-infected cells that can bind TCR, irrespective of its antigenic specificity, resulting in the activation of a large number of T lymphocytes of different antigenic specificity, thus behaving as a potent immune-stimulating molecule [18].
Bacterial superantigens bind to MHC molecules outside of their peptide binding grooves and interact predominantly with only the Vβ domains of TCRs, resulting in the stimulation of up to 20 percent of the entire T cell population. In this way, SAGs initiate a systemic release of inflammatory cytokines that results in various immune-mediated diseases. Bacterial superantigens have also been implicated in the pathogenesis of arthritis, asthma and inflammatory bowel syndrome [19].
Microchimerism
A longer term effect of pregnancy is the persistence of fetal cells in women after a pregnancy and of maternal cells in her offspring, known as microchimerism. These cells can be hematopoietic or can differentiate into somatic cells in multiple organs and are found in both healthy individuals and those with autoimmune diseases. How these cells are tolerated by the immune system is poorly understood, but it is possible that if these cells are targeted as foreign cells they could be implicated in the pathogenesis of autoimmune diseases [20].
In an article, microchimerism in peripheral blood mononuclear cells has been shown to be more frequent in women with scleroderma than healthy controls [21]. Another study has also shown that microchimerism with male DNA is also found in women who have never given birth to a son and suggests other sources of DNA, not only a history of a male birth, must be used in research studies [22]. Overall results indicate that fetal microchimerism is a common phenomenon including in healthy women, but that microchimerism levels are increased in women with systemic sclerosis [23].
A role for fetal microchimerism in systemic lupus erythematosus has also been controversial [24], although several studies suggest that lupus nephritis in particular is associated with an increased concentration of fetal microchimerism in circulation and in renal tissue [25, 26].
Within maternal tissues, the fetal microchimeric progenitor immature T cells, also known as CD4 cells, are capable of self-renewal, proliferation, differentiation and activation. Activation of progenitor cells can result in the production of paracrine and autocrine inflammatory cytokines and chemokines that are involved in autoimmune diseases [27]. Animal experimentation and collection of human data will be necessary to understand the relationship between fetal microchimerism and specific autoimmune diseases in women.
Can infections because of autoimmune diseases?
Infections and autoimmune diseases have multifaceted and multidirectional relationships [28].
Recently, it has been considered that infections cannot only induce or precipitate autoimmune diseases, but they may also protect from autoimmunity or even abrogate an ongoing autoimmune process depending on the interaction between microorganisms and host [29]. Viruses and bacteria are the infectious agents that have been long associated with autoimmune diseases. Some of these agents and diseases will be discussed.
Cytomegalovirus
Cytomegalovirus (CMV) infects 70–100 % of adults in populations worldwide [29]. The development of systemic lupus erythematosus (SLE) may be triggered by a CMV infection. Existing SLE may undergo an exacerbation following a CMV infection [30]. Serological signs of active CMV infection have been detected in SLE patients [31]. In addition, the infection is associated with increased disease activity [32]. The virus has been detected in patients with RA, Sjögren’s syndrome, dermatopolymyositis, psoriasis, Wegener’s granulomatosis, ulcerative colitis and Crohn’s disease [29].
Epstein–Barr virus
Another agent searched for pathogenesis of autoimmune diseases is Epstein–Barr virus (EBV). EBV is a human DNA virus that infects B cells and causes their polyclonal activation and produces polyclonal antibodies. Polyclonal B cell activation may be an early step in the pathogenesis of SLE. Serologic association, cross-reactivity of select EBV specific antibodies with SLE autoantigens, SLE-like autoimmunity after immunization with EBV peptides, increased viral load in SLE patients and SLE-specific alterations in EBV humoral and cellular immunity include EBV in the development of SLE [33, 34]. Esen et al. [35] reported significantly higher early antigen (EA/D) IgG immune response in SLE patients. The authors informed that the serologic profile of patients in the study may indicate reactivation of EBV infection in SLE patients which may be due to immune dysregulation induced by the disease itself, the effect of immunosuppressive therapy or the result of molecular mimicry mechanisms. In another study by Zandman-Goddard et al. [36], anti-EA IgG antibodies were found to be higher in lupus patients with cutaneous and joint manifestations and increased anti-Ro antibody titers.
Parvovirus B19
Lunardi et al. [37] have recently determined a peptide that shares homology with the capsid protein VP1 of Parvovirus and with human cytokeratin. Supporting the molecular mimicry hypothesis in the pathogenesis of autoimmune diseases, this peptide also shares similarity with globulin transcription factor 1, which plays a significant role in megakaryopoiesis and in erythropoiesis. Endothelial cell damage may also be related to Parvovirus infections. A direct correlation between the extent of degenerative endothelial cell alterations and the degree of B19 RNA expression suggested a causal role of B19 in the propagation of the endothelial cell dysfunction [38].
In addition, chronic Parvovirus B19 infection can induce antiviral antibodies that also react specifically with collagen type II, single-stranded DNA and cardiolipin (29).
Zakrzewska et al. [39] studied Parvovirus B19 in SSc patients and showed some differences in the rate of persistence of B19 V DNA, in the simultaneous persistence of two genotypes and in the pattern of viral expression among SSc patients and controls.
Hepatitis C Virus
Ramos-Casals et al. [40] have investigated the clinical and immunologic characteristics of a large series of patients with systemic autoimmune diseases associated with chronic hepatitis C virus (HCV) infection. In the study group, Sjögren’s syndrome (SS), RA and SLE were associated with chronic HCV infection.
In another study [41], it has been emphasized the strong associations of autoimmune thyroid disease with the HCV infection and interferon-α (IFNα) therapy. In addition, it was likely that HCV and IFN act in synergism to trigger autoimmune thyroid disease in patients. The association between HCV infection and thyroid autoimmunity and type 2 diabetes mellitus has been reported, and a common pathogenetic pathway of HCV-related extrahepatic diseases with these autoimmune endocrine manifestations has been suggested [42]. Chronic antigenic stimulation by HCV is considered a key mechanism sustaining the proliferation of rheumatoid factor-secreting B cell clones. It has been hypothesized that it may play a role as an early, chronic stimulus for autoreactive B cells in HCV-infected patients [43].
Hepatitis B Virus
Hepatitis B virus is associated with liver disease, but is also related to extrahepatic manifestations, such as prodromal serum sickness in acute hepatitis, membranous glomerulonephritis, membranoproliferative glomerulonephritis, cutaneous vasculitis, infantile popular acrodermatitis, essential mixed cryoglobulinaemia and polyarteritis nodosa (PAN), all forms of immune complex diseases. Furthermore, HBV infection is associated with other inflammatory syndromes in diseases such as rheumatoid arthritis, polymyalgia rheumatica and polymyositis [44, 45].
Several mechanisms have been linked to HBV as the inducer of some autoimmune phenomena. The mechanisms include: molecular mimicry between HBV antigens and self proteins, the generation of immune complexes between HBV antigens and antibodies and apoptosis/tissue damage resulting in the exposure of intracellular antigens [45, 46].
Influenza
Sivadon-Tardy et al. [47] reported that influenza virus can also induce Guillain–Barre′ syndrome, but the majority of infections were due to virus A (H3N2). However, Chaari et al. [48] have informed a case of Guillain–Barre′ syndrome (GBS) related to pandemic influenza A (H1N1) infection. In another article, it was informed that an analysis was conducted of 10,486 acute flaccid paralysis cases diagnosed as Guillain–Barre′ syndrome from 2000 through 2008 in children aged <15 years in Latin American and the Caribbean countries and territories. The acute flaccid paralysis surveillance system have represented a useful means of monitoring GBS during the pandemic [49].
Campylobacter
Drenthen et al. [50] discussed retrospective analysis of preceding infections in relation to serial electrophysiology and clinical data from 123 GBS patients. Accordingly, 17 (14 %) of 123 patients had C. jejuni-related GBS. C. jejuni patients had lower motor and higher sensory action potentials compared with viral-related cases. In another study, Usuki et al. [51] have established a rat model of peripheral nerve dysfunction induced by antiganglioside antibodies via sensitization by the lipooligosaccharide of C. jejuni. They have shown further that it is possible to utilize an anti-idiotype antibody design, on the basis of molecular mimicry, to ameliorate the neurological dysfunction in this animal model.
Streptococcus pyogenes
Guilherme and Kalil [52], using a proteomics approach, identified myocardium and valvular autoantigens that were recognized by heart-infiltrating and peripheral T cells from rheumatic fever/chronic rheumatic heart disease (RF/RHD) patients. Their results showed that there were several expanded T cell populations with an oligoclonal profile in the heart tissue of chronic and acute RHD patients. The researchers emphasized that molecular mimicry is defined as a sharing of epitopes between antigens of the host and the infectious agent, which in the case of RF/RHD is S. pyogenes.
In patients with Sydenham chorea antibody, cross-reactivity between S. pyogenes membrane and neuronal cytoplasm has been reported [29].
Staphylococcus aureus
Mulvey et al. [53] examined the association between individuals with multiple sclerosis (MS) and colonization with S. aureus harboring superantigens. In the research, nasal swabs were collected from non-MS subjects and patients with MS who had not experienced a relapse in the past 6 months (MS stable group) and who had suffered a relapse within 30 days of study recruitment (MS exacerbation group). They informed that among individuals colonized with S. aureus, the prevalence of staphylococcal superantigen gene (sea) was significantly greater in the MS exacerbation versus non-MS study group. This issue has been discussed in variety of articles frequently [54, 55].
Genetics, autoimmunity and infection
Genetics play an important role in autoimmunity and influence the response of an individual to environmental factors such as infections. The interaction among genetics, infection and autoimmunity was studied in a series of animal experiments by investigators. Neu et al. [56] demonstrated that infection with coxsackievirus induced autoimmune myocarditis in genetically susceptible BALB/c mice, dependent on major histocompatibility complex (MHC) and non-MHC genes. In humans, the presence of human leukocyte antigen (HLA) DR11 phenotype was linked to mixed cryoglobulinemia (MC) in patients with chronic HCV infection. In contrast, HLA-DR7 seemed to protect HCV-infected patients from mixed cryoglobulinemia [57]. Kudat et al. [58] informed that some HLA class II DR and DQ alleles such as HLA-DRB1*07 were found to be associated with the risk of developing post-Streptococcus acute rheumatic fever (RF). Furthermore, the HLA-DRB1*13, DRB5* and DRB3* were protective against the development of rheumatic valve damage. HLA-DR2 is associated with MS [59] and SLE [60], and HLA-B27 with ankylosing spondylitis (AS) [61]. Meanwhile, the frequency of HLA-B27 allelotypes in Crohn’s disease (CD) patients without associated arthritis is usually the same as in the normal population, but it is increased to up to 60 % in those with involvement of the spinal joints [62]. In rheumatoid arthritis (RA), class II MHC gene, HLA-DR4, is the most strongly linked genetic marker to this disease. The frequency of this allelotype has been found to be around 70 % in RA patients, but it is detected in less than 30 % of the general population [63].
Vulnerability to infections can be influenced by genetic profiles. For instance, black ethnic groups are more prone to infection with mycobacterium tuberculosis and meningococcemia than are Caucasians [64].
Since the mid 1980 s, many studies have emphasized a role of Proteus mirabilis in the etiopathogenesis of RA [65]. For instance, Ebringer et al. [66] informed that rabbits injected with HLA-DR4-positive lymphocytes were found to produce antibodies which will only bind to P. mirabilis but not 18 other microorganisms. In another study, tissue-typing sera from pregnant women having anti-HLA-DR4 specificity were found to bind more significantly to P. mirabilis than to E. coli [67]. IgG antibodies from patients with RA were found to have cytotoxic activities against HLA-DR4 cells as shown by increased hemolysis for the sheep red blood cells coated with HLA-DRβ1*0404 peptides when compared to sera from AS and healthy control subjects [68]. These findings support the notions that there is a significant role for Proteus microorganisms in the initiation and perpetuation of RA.
The results of various studies have indicated microbiological evidence for a link between Klebsiella microorganisms and AS. Anti-sera from immunized rabbits with Klebsiella were found to bind equally to HLA-B27-positive lymphocytes whether obtained from AS patients or healthy controls but not to lymphocytes taken from HLA-B27-negative individuals [69]. Mäki-Ikola et al. [70] informed that Klebsiella antibodies were significantly elevated in the serum compared to the synovial fluid of AS patients. The result indicates that these antibodies are produced in extra-articular regions such as the enteric mucosal lymphatic system before gaining entry into the joints.
As can be inferred from the studies, genetic susceptibility might explain why only a subgroup of individuals will develop autoimmunity after infections. The relationship between immune dysregulation and autoimmune disease that can be triggered by an infectious agent has been demonstrated in animal models such as New Zealand black/white (NZB/W) mice and non-obese diabetic (NOD) mice. NZB/W mice are genetically prone to develop an SLE-like disease, exhibited by autoimmune hemolytic anemia, nephritis and high resistance to induction of tolerance [64].
Is there any protective feature of infections in autoimmune diseases?
The effects of certain parasitic and bacterial infections are to moderate the immune response; an excessive response can have a pathological outcome [71]. Some studies using animal models have led to the suggestion that human autoimmune or allergic diseases might be alleviated by the use of microbial products. There are some data that would support such an observation [72]. For example, it was demonstrated that infection with Schistosoma mansoni [73] or Coxsackieviruses [74] can prevent diabetes in NOD mice. This protective effect is result of a strong Th2- and regulatory T cell response [73]. Krause et al. [75] also demonstrated epidemiological support for the protective role of infections. They found significantly fewer antibodies against H. pylori, CMV, EBV and Toxoplasma in sera of Type 1 diabetes (T1D) patients compared with their first-degree family members or healthy controls. Ram et al. [76] found a lower prevalence of anti-hepatitis B antibodies among patients with MS and SLE.
According to the “Hygiene Hypothesis”, Strachan [77] assumed that the increase in ADs observed in Western countries was partly caused by a decline in infectious diseases and improved hygiene. In some cases, infections can actually protect individuals from autoimmune and allergic diseases. It was suggested that reduced exposure to infectious agents in infancy might predispose to hay fever. “Hygiene Hypothesis” applies to most autoimmune diseases, especially multiple sclerosis [78] and T1D [79].
Conclusion
There is an obvious fact that relationships between infections and many autoimmune diseases are complicated. Infections may trigger autoimmunity, and many different infectious agents seem to be potentially involved in the induction of different autoimmune diseases. The etiopathogenetic mechanism which plays a major role in the causation and the development of some autoimmune diseases involves interplay between the genetic and environmental factors. However, microorganisms form an important part of causal connection in the most immune-mediated rheumatic diseases.
References
National Institutes of Health (2002) Autoimmune disease coordinating committee report 2002. The Institutes, Bethesda (MD)
Rose NR (2002) An immunology primer. In: Morton CC, Fagan T (eds) Proceedings from sex differences in immunology and autoimmunity, Society for Women’s Health Research, Boston, MA, 8 Nov 2001. Society for Women’s Health Research, Washington, pp 7–9
Rose NR (2002) Mechanisms of autoimmunity. Semin Liver Dis 22:387–394
Kamradt T (2005) Can infection prevent or cure allergy and autoimmunity? Discov Med 5:283–287
Grossman C, Dovrish Z, Shoenfeld Y, Amital H (2011) Do infections facilitate the emergence of systemic sclerosis? Autoimmune Rev 10:244–247
Grigoriadis N, Hadjigeorgiou GM (2006) Virus-mediated autoimmunity in multiple sclerosis. J Autoimmune Dis. doi:10.1186/1740-2557-3-1
Francis L, Pearl A (2010) Infection in systemic lupus erythematosus: friend or foe? Int J Clin Rheumtol 5:59–74
Olson JK, Ludovic Croxford J, Miller SD (2004) Innate and adaptive immune requirements for induction of autoimmune demyelinating disease by molecular mimicry. Mol Immunol 40:1103–1108
Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A (2005) Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol Rev 204:27–42
Rahman AH, Eisenberg RA (2006) The role of toll-like receptors in systemic lupus erythematosus. Semin Immunopathol 28:131–143
Richez C, Blanco P, Rifkin I, Moreau JF, Schaeverbeke T (2011) Role for toll-like receptors in autoimmune disease: the example of systemic lupus erythematosus. Joint Bone Spine 78:124–130
Kerbel R, Folkman J (2002) Clinical translation of angiogenesis inhibitors. Nat Rev Cancer 2:727–739
Guiducci S, Distler O, Distler JHW, Matucci-Cerinic M (2008) Mechanisms of vascular damage in SSc-implications for vascular treatment strategies. Rheumatology 47:18–20
Haskard DO (1995) Cell adhesion molecules in rheumatoid arthritis. Curr Opin Rheumatol 7:229–234
Veale DJ, Maple C, Kirk G, McLaren M, Belch JJ (1998) Soluble cell adhesion molecules: P-selecting and ICAM-1, and disease activity in patients receiving sulphasalazine for active rheumatoid artritis. Scand J Rheumatol 27:296–299
Ku IA, Imboden JB, Hsue PY, Ganz P (2009) Rheumatoid arthritis-A model of systemic inflammation driving atherosclerosis. Circ J 73:977–985
Sgonc R, Gruschwitz MS, Dietrich H, Recheis H, Gershwin ME, Wick G (1996) Endothelial cell apoptosis is a primary pathogenetic event underlying skin lesions in avian and human scleroderma. J Clin Invest 98:785–792
Samarkos M, Vaiopoulos G (2005) The role of infections in the pathogenesis of autoimmune diseases. Curr Drug Targets Inflamm Allergy 4:99–103
Sundberg EJ, Deng L, Mariuzza RA (2007) TCR recognition of peptide/MHC class II complexes and superantigens. Semin Immunol 19:262–271
Oliver JE, Silman AJ (2009) Why are women predisposed to autoimmune rheumatic diseases? Arthritis Res Ther. doi:10.1186/ar2825
Evans PC, Lambert N, Maloney S, Furst DE, Moore JM, Nelson JL (1999) Long-term fetal microchimerism in peripheral blood mononuclear cell subsets in healthy women and women with scleroderma. Blood 93:2033–2037
Lambert NC, Pang JM, Yan Z, Erickson TD, Stevens AM, Furst DE, Nelson JL (2005) Male microchimerism in women with systemic sclerosis and healthy women who have never given birth to a son. Ann Rheum Dis 64:845–848
Gammill HS, Nelson JL (2010) Naturally acquired microchimerism. Int J Dev Biol 54:531–543
Khosrotehrani K, Mery L, Aractingi S, Bianchi DW, Johnson KL (2005) Absence of fetal cell microchimerism in cutaneus lesions of lupus erythematosus. Ann Rheum Dis 64:159–160
Mosca M, Curcio M, Lapi S, Valentini G, D’angelo S, Rizzo G, Bombardieri S (2003) Correlations of Y chromosome microchimerism with disease activity in patients with SLE: analysis of preliminary data. Ann Rheum Dis 62:651–654
Hovinga ICLK, Koopmans M, Baelde HJ, Vanderwal AM, Sijpkens YWJ, Deheer E, Bruijn JA, Bajema IM (2006) Chimerism occurs twice as often in lupus nephritis as in normal kidneys. Arthritis Rheum 54:2944–2950
Miech RP (2010) The role of microchimerism in autoimmune disease. Int J Clin Exp Med 3:164–168
Doria A, Sarzi-Puttini P, Shoenfeld Y (2008) Infections, rheumatism and autoimmunity: the conflicting relationship between humans and their environment. Autoimmune Rev 8:1–4
Sfriso P, Ghirardello A, Botsios C, Tonon M, Magherita Z, Bassi N, Bassetto F, Dorio A (2010) Infections and autoimmunity: the multifaced relationship. J Leukoc Biol 87:385–395
Goddard GZ, Shoenfeld Y (2005) Infections and SLE. Autoimmunity 38:473–485
Barzilai O, Sherer Y, Ram M, Izhaky D, Anaya JM, Shoenfeld Y (2007) Ebstein-Barr virus and cytomegalovirus in autoimmune diseases:are they truly notorious? A preliminary report. Ann N Y Acad Sci 1108:567–577
Su BY, Su CY, Yu SF, Chen CJ (2007) Incidental discovery of high systemic lupus erythematosus disease activity associated with cytomegalovirus viral activity. Med Microbiol Immunol 196:165–170
Toussirrot E (2008) Epstein Barr virus in autoimmune disease. Best Pract Res Clin Rheumatol 22:883–896
James JA, Robertson JM (2012) Lupus and Epstein-Barr. Curr Opin Rheumatol. doi:10.1097/BOR.0b013e3283535801
Esen BA, Yılmaz G, Uzun S, Ozdamar M, Aksozek A, Kamalı S, Turkoglu S, Gulş A, Ocal L, Aral O, Inanc M (2010) Serologic response to Epstein-Barr virus antigens in patients with systemic lupus erythematosus: a controlled study. Rheumatol Int. doi:10.1007/s00296-010-1573-4
Zandman-Goddard G, Berkun Y, Barzilai O, Boaz M, Blank M, Ram M, Sherer Y, Anaya JM, Shoenfeld Y (2009) Exposure to Epstein-Barr virus infection is associated with mild systemic lupus erythematosus disease. Ann NY Acad Sci 1173:658–663
Lunardi C, Tinazzi E, Bason C, Dolcino M, Corrocher R, Puccetti A (2008) Human parvovirus B19 infection and autoimmunity. Autoimmune Rev 8:116–120
Magro CM, Nuovo GJ, Ferri C, Crowson AN, Giuggioli D, Sebastiani M (2004) Parvoviral infection of endothelial cells and stromal fibroblasts: a possible pathogenetic role in scleroderma. J Cutan Pathol 31:43–50
Zakrzewska K, Corcioli F, Carlsen KM, Giuggioli D, Fanci R, Rinieri A, Ferri C, Azzi A (2009) Human parvovirus B19 (B19 V) infection in systemic sclerosis patients. Intervirology 52:279–282
Ramos-Casals M, Muñoz S, Medina F, Jara LJ, Rosas J, Calvo-Alen J, Brito-Zerón P, Forns X, Sánchez-Tapias JM; HISPAMEC Study Group (2009) Systemic autoimmune diseases in patients with hepatitis C virus infection: characterization of 1020 cases (The HISPAMEC registry). J Rheumatol 36:1442–1448
Menconi F, Hahsam A, Tomer Y (2011) Environmental triggers of thyroiditis: hepatitis C and interferon-α. J Endocrinol Invest 34:78–84
Antonelli A, Ferri C, Ferrari SM, Colaci M, Fallahi P (2008) Immunpathogenesis of HCV related endocrine manifestations in chronic hepatitis and mixed cryoglobulinemia. Autoimmune Rev 8:18–23
De Vita S, Quartuccio L, Fabris M (2008) Hepatitis C virus infection, mixed cryoglobulinemia and BlyS upregulation: targeting the infectious trigger, the autoimmune response, or both? Autoimmune Rev 8:95–99
Shepard CW, Simard EP, Finelli L, Fiore AE, Bell BP (2006) Hepatitis B virus infection: epidemiology and vaccination. Epidemiol Rev 28:112–125
Maya R, Gershwin ME, Shoenfeld Y (2008) Hepatitis B Virus (HBV) and autoimmune disease. Clinic Rev Allerg Immunol 34:85–102
Chi ZC, Ma SZ (2003) Rheumatologic manifestations of hepatic diseases. Hepatobiliary Pancreat Dis Int 2:32–37
Sivadon-Tardy V, Orlikowski D, Porcher R, Sharshar T, Durand MC, Enouf V, Rozenberg F, Caudie C, Annane D, van der Werf S, Lebon P, Raphael JC, Gaillard JL, Gault E (2009) Guillain–Barre′ syndrome and influenza virus infection. Clin Infect Dis 1:48–56
Chaari A, Bahloul M, Dammak H, Nourhene G, Rekik N, Hedi C, Chokri BH, Bouaziz M (2010) Guillain-Barré syndrome related to pandemic influenza A (H1N1) infection. Intensive Care Med 36:1275
Landaverde JM, Danovaro-Holliday MC, Trumbo SP, Pacis-Tirso CL, Ruiz-Matus C (2010) Guillain-Barré syndrome in children aged < 15 years in Latin America and the Caribbean: baseline rates in the context of the influenza A (H1N1) pandemic. J Infect Dis 201:746–750
Drenthen J, Yuki N, Meulstee J, Maathuis EM, van Doorn PA, Visser GH, Blok JH, Jacobs BC (2011) Guillain-Barré’ syndrome subtypes related to Campylobacter infection. J Neurol Neurosurg Psychiatry 82:300–305
Usuki S, Taguchi K, Thompson SA, Chapman PB, Yu RK (2010) Novel anti-idiotype antibody therapy for lipooligosaccharide-induced experimental autoimmune neuritis: use relevant to Guillain-Barré syndrome. J Neurosci Res 88:1651–1663
Guilherme L, Kalil J (2010) Mechanisms leading autoimmune reactivity and disease. J Clin Immunol 30:17–23
Mulvey MR, Doupe M, Prout M, Leong C, Hizon R, Grossberndt A, Klowak M, Gupta A, Melanson M, Gomori A, Esfahani F, Klassen L, Frost EE, Namaka M (2011) Staphylococcus aureus harbouring enterotoxin A as a possible risk factor for multiple sclerosis exacerbations. Mult Scler 17:397–403
Martin E, Winn R, Nugent K (2011) Catastrophic antiphospholipid syndrome in a community-acquired methicillin-resistant Staphylococcus aureus infection: a review of pathogenesis with a case for molecular mimicry. Autoimmune Rev 10:181–188
Laudien M, Gadola SD, Podschun R, Hedderich J, Paulsen J, Reinhold-Keller E, Csernok E, Ambrosch P, Hellmich B, Moosig F, Gross WL, Sahly H, Lamprecht P (2010) Nasal carriage of Staphylococcus aureus and endonasal activity in Wegeners granulomatosis as compared to rheumatoid arthritis and chronic rhinosinusitis with nasal polyps. Clin Exp Rheumatol 28(1 Suppl 57):51–55
Neu N, Rose NR, Beisel KW, Herskowitz A, Gurri-Glass G, Craig SW (1987) Cardiac myosin induces myocarditis in genetically predisposed mice. J Immunol 139:3630–3636
Cacoub P, Renou C, Kerr G, Hüe S, Rosenthal E, Cohen P, Kaplanski G, Charlotte F, Thibault V, Ghillani P, Piette JC, Caillat-Zucman S (2001) Influence of HLA-DR phenotype on the risk of hepatitis C virus-associated mixed cryoglobulinemia. Arthritis Rheum 44:2118–2124
Kudat H, Telci G, Sozen AB, Oguz F, Akkaya V, Ozcan M, Atilgan D, Carin M, Guven O (2006) The role of HLA molecules in susceptibility to chronic rheumatic heart disease. Int J Immunogenet 33:41–44
Haines JL, Terwedow HA, Burgess K, Pericak-Vance MA, Rimmler JB, Martin ER, Oksenberg JR, Lincoln R, Zhang DY, Banatao DR, Gatto N, Goodkin DE, Hauser SL et al (1998) Linkage of the MHC to familial multiple sclerosis suggests genetic heterogeneity. The Multiple Sclerosis Genetics Group. Hum Mol Genet 7:1229–1234
Graham RR, Ortmann W, Rodine P, Espe K, Langefeld C, Lange E, Williams A, Beck S, Kyogoku C, Moser K, Gaffney P, Gregersen PK, Criswell LA, Harley JB, Behrens TW (2007) Specific combinations of HLA-DR2 and DR3 class II haplotypes contribute graded risk for disease susceptibility and autoantibodies in human SLE. Eur J Hum Genet 15:823–830
van der Linden SM, Valkenburg HA, de Jongh BM, Cats A (1984) The risk of developing ankylosing spondylitis in HLA-B27 positive individuals. A comparison of relatives of spondylitis patients with the general population. Arthritis Rheum 27:241–249
Braun J, Sieper J (2010) Ankylosing spondylitis, other spondyloarthritides, and related conditions. In: Warrell DA, Cox TM, Firth JD (eds) Oxford textbook of medicine. Oxford University Press, Oxford, pp 3603–3616
Stastny P (1978) Association of the B-cell alloantigen DRw4 with rheumatoid arthritis. N Engl J Med 298:869–871
Kivity S, Agmon-Levin N, Blank M, Shoenfeld Y (2009) Infections and autoimmunity-friends or foes? Trends Immunol 30:409–414
Rashid T, Ebringer A (2012) Autoimmunity in rheumatic diseases is induced by microbial infections via crossreactivity or molecular mimicry. Autoimmune Dis. doi:10.1155/2012/539282
Ebringer A, Ptaszynska T, Corbett M (1985) Antibodies to Proteus in rheumatoid arthritis. Lancet 2:305–307
Khalafpour S, Ebringer A (1987) Cross-reactivity between HLA-DR4 and Proteus mirabilis. Periodic Biol (Zagreb) 89(suppl. 1):203
Wilson C, Rashid T, Tiwana H, Beyan H, Hughes L, Bansal S, Ebringer A, Binder A (2003) Cytotoxicity responses to peptide antigens in rheumatoid arthritis and ankylosing spondylitis. J Rheumatol 30:972–978
Baines M, Ebringer A, Avakian H, Samuel D, James DCO (1990) The use of enzyme immunoassay (EIA) and radiobinding assay to investigate the cross-reactivity of Klebsiella antigens and HLAB27 in ankylosing spondylitis patients and healthy controls. Scand J Rheumatol 19:341–349
Mäki-Ikola O, Penttinen M, Von Essen R, Gripenberg-Lerche C, Isomäki H, Granfors K (1997) IgM, IgG and IgA class enterobacterial antibodies in serum and synovial fluid in patients with ankylosing spondylitis and rheumatoid arthritis. Br J Rheumatol 36:1051–1053
Cooke A (2009) Infection and autoimmunity. Blood Cells Mol Dis 42:105–107
Gaisford W, Cooke A (2009) Can infections protect against autoimmunity? Curr Opin Rheumatol 21:391–396
Zaccone P, Fehérvári Z, Jones FM, Sidobre S, Kronenberg M, Dunne DW, Cooke A (2003) Schistosoma mansoni antigens modulate the activity of the innate immuneresponse and prevent onset of type 1 diabetes. Eur J Immunol 33:1439–1449
Tracy S, Drescher KM, Chapman NM, Kim KS, Carson SD, Pirruccello S, Lane PH, Romero JR, Leser JS (2002) Toward testing the hypothesis that group B coxsackieviruses (CVB) trigger insulin-dependent diabetes: inoculating nonobese diabetic mice with CVBmarkedly lowers diabetes incidence. J Virol 76:12097–12111
Krause I, Anaya JM, Fraser A, Barzilai O, Ram M, Abad V, Arango A, García J, Shoenfeld Y (2009) Anti-infectious antibodies and autoimmune-associated autoantibodies inpatients with type I diabetes mellitus and their close family members. Ann N Y Acad Sci 1173:633–639
Ram M, Anaya JM, Barzilai O, Izhaky D, Porat Katz BS, Blank M, Shoenfeld Y (2008) The putative protective rol of hepatitis B virus (HBV) infection from autoimmune disorders. Autoimmune Rev 7:621–625
Strachan DP (1989) Hay fever, hygiene, and household size. BMJ 299:1259–1260
Pugliatti M, Sotgiu S, Rosati G (2002) The worldwide prevalence of multiple sclerosis. Clin Neurol Neurosurg 104:182–191
Gale EA (2002) The rise of childhood type 1 diabetes in the 20th century. Diabetes 51:3353–3361
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sener, A.G., Afsar, I. Infection and autoimmune disease. Rheumatol Int 32, 3331–3338 (2012). https://doi.org/10.1007/s00296-012-2451-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00296-012-2451-z