
Abstract
Analysis of virulence mechanisms of plant pathogens is often limited by the lack of genetic tools that can be used to identify genes that are preferentially expressed during their interactions with plants. In the present study, we used the newly constructed IVET (in vivo expression technique) plasmid pIviGK and the corresponding antibiotic resistance–based selection method to identify genes that encode pathogenicity factors of the soft rot-causing bacterium Pseudomonas viridiflava. These included pel, the gene encoding pectate lyase, which is responsible for the development of soft rot symptoms. We have also isolated and characterized the gene mviN pv encoding a putative novel membrane associated virulence factor of P. viridiflava. A mutation in mviN pv was shown to influence motility as well as virulence of P. viridiflava. The mviN pv gene is expressed to a moderate level in LB media and its expression increases under inducing conditions as was shown by measuring in planta expression dynamics of the fused gfp reporter gene.
Similar content being viewed by others

The genetic determinants of bacterial virulence are tightly and precisely regulated, mainly at the level of transcription. This allows isolating pathogenicity-associated genes based on their specific in vivo (in planta) expression. The in vivo expression technique (IVET) [12], a promoter probing method, is one of the approaches that allows the positive selection of mutants for the identification of genes that are specifically fused with promoterless reporter genes. The end result of this analyses is the identification of bacterial genes that are expressed preferentially during conditions relevant to the disease process. Several studies report the application of IVET in different animal–bacterial interactions [2]. In addition, this method has been used successfully to study in vivo bacterial gene activation under different environmental conditions, such as during endophytic colonization of plants [15, 21], bacterial growth in soil [24] or more recently, plant invasion of phytopathogens [3, 18].
Here we report the construction of two promoter probing plasmids, pIviGK and pIviGG, which contain gfp and kanamycin or gentamicin resistance reporter gene pairs, respectively. We successfully used pIviGK to identify several genes of Pseudomonas viridiflava that are induced during the infection of pepper fruit. Thereby, it has been validated that this plasmid is applicable for the isolation of bacterial genes induced under specific (in planta) conditions. One such gene encodes a homologue of the MviN membrane protein of Salmonella typhimurium [4] and a gene encoding pectate lyase (Pel, EC 4.2.2.2) responsible for the development of soft rot symptoms [10]. In this study, we have examined whether the MviN homologue contributed only to the motility of P. viridiflava or also to its virulence. Finally, fluorometric measurements, based on activity of the gfp reporter gene, were used to study in planta expression dynamics of pel and mviN pv .
Materials and Methods
Bacterial strains and plasmids
Bacteria used in this study were: Pseudomonas viridiflava, Escherichia coli DH5α/λ pir, E. coli S17.1, and E. coli HB101. Bacterial cultures were maintained and cultivated on Luria Bertani (LB) media. Antibiotics for selection were: 50 μg/mL rifampicin, 10 μg/mL gentamicin, and 50 μg/mL kanamycin. Plasmids used in this study were: pRK2013 [6] pBluescriptKS (Stratagene, La Jolla, CA), pGP704 [11], pJQ199 [19], and pAG408 [25].
Recombinant DNA techniques
Plasmid DNA isolation, transformation of E. coli, agarose gel electrophoresis, and all enzymatic treatments were conducted as described by Sambrook et al. [22]. DNA fragments were isolated from agarose gels using GELase (EPICENTRE) following the manufacturer’s recommendations. P. viridiflava total genomic DNA was isolated according to Pitcher et al. [17].
Construction of pIviGK and pIviGG
A derivative of pGP704, designated pGPMCSII, which contains the MCSII synthetic polylinker sequence (Fig. 1) between the BglII and KpnI sites, was created. The gentamicin (accA1) and kanamycin resistance genes (aphA3) were isolated from pAG408. The ends of both isolated fragments were blunted with T4 polymerase and ligated to SspI and Bst1107I double-digested pGPMCSII, resulting in pGPMCSII: gm and pGPMCSII: km. Gfp-km or gfp-gm reporter gene pairs were created as follows. Promoterless derivatives of aphA3 and accA1 genes (afterwards: kmS/R and gmS/R, respectively) were created by nested PCR. Products of the PCR reactions were digested with EcoRI and SmaI and ligated to pBluescriptKS resulting in pBKS:gmS/R and pBKS:kmS/R, respectively. These plasmids were digested with KpnI and EcoRI and ligated to the atpE-gfp gene cassette originated from pAG408, resulting in pBKS: gfp-gmS/R and pBKS:gfp-kmS/R. Reporter gene pairs were isolated by KpnI and SmaI double digestions and were ligated to pGPMCSII:km or pGPMCSII:gm, resulting in pIviGK and pIviGG plasmids.

Physical map of pIviGK. The MCS (multi-cloning site) and the flanking region that contains the PacI site on both sides, and the pUC/M13 sequencing primer recognition sites, are shown enlarged (top panel). The TIS (translation initiation sequence) contains the 5′-TTAACTTTA epsilon translational enhancer of gene 10 of bacteriophage T7 [14] along with the 5′-TGATCC translation–initiation promoting site (TPS) [26] that invariably exists in the mRNA of highly expressed bacterial genes. The three-way translational stop codons are underlined in the sequences.
Construction of the Ivi-gene fusion library
Purified P. viridiflava genomic DNA was digested with HindIII, fragments of 2,000–3,000 bp were isolated from gel. The 5′-overhanging ends of the isolated fragments were partially backfilled with dATP and dGTP using Klenow DNA polymerase. XbaI digested pIviGK that was partially filled with dCTP and dTTP was ligated to P. viridiflava genomic DNA fragments. All (approximately 104) Escherichia coli DH5α/λ pir transformants were used as a donor in a conjugation reaction wherein P. viridiflava was the recipient.
Infiltration of green pepper fruits
A 30-sec vacuum infiltration method was used for the infection of 0.5-cm-wide green pepper rings with P. viridiflava suspensions.
In vivo selection
The IVET–mutant P. viridiflava pool was created from 104 colonies and suspensions were used for vacuum infiltration. Two hours post infection, pepper rings were vacuum infiltrated with 50 μg/ml kanamycin solution. After 10–12 hours of incubation, the pepper rings were homogenated and bacteria were suspended and plated onto selective LB media containing gentamicin and incubated for 48 hours at 28°C. Petri dishes were exposed shortly to UV irradiation using a UV transilluminator and colonies lacking green light emission were observed by visual scoring and picked for further characterisation.
Conjugative cloning and sequencing of ivi-gene fusions
Plasmids pIviGK with inserts of different ivi genes were mobilised into E. coli DH5α/λ pir by retrotransfer [20]. ivi genes were sequenced using the pUC/M13 (-26) sequencing primer.
Infiltration of Arabidopsis leaves and assessment of bacterial multiplication in planta
The infection of Arabidopsis thaliana (ecotype Col-1) leaves using a blunt-end syringe or the dipping inoculation method along with the enumeration of bacteria were carried out using an established method [7].
Measuring expression dynamics of ivi genes by fluorescence
Fluorescence measurements were carried out on a Fluoromax-3 (Jobin Yvon, France) spectrofluorometer. P. viridiflava cells conditionally expressing GFP were excited at 395 nm. Fluorescent emission was detected at 512 nm. P. viridiflava ivi-mutants were grown overnight in liquid LB media supplemented with 10 μg/ml gentamicin. Bacteria were centrifuged and pellets resuspended in distilled water and pepper rings were vacuum infiltrated. Pepper rings were pulped at different times after infiltration and fluorescence was measured.
Creating PV-dm, an mviN pv knockout mutant of P. viridiflava
The mviN pv containing chromosomal fragment was amplified by PCR from pIviGK::s1/6. The ends of the PCR product were back-filled using T4 polymerase and cloned into the unique SmaI site of pJQ199. A kanamycin resistance cassette was inserted into the unique SmaI site of mviN pv and the construction was transformed into the donor E. coli S17.1. The mutated mviN pv was marker-exchanged as described previously [19] into P. viridiflava to construct strain PV-dm. The correct insertion of the aphA3 cassette was verified by colony PCR.
Nucleotide sequence accession numbers
The nucleotide sequences reported in this paper have been deposited in GenBank under accession numbers DQ077710 (MCSII of pIviGK) and DQ077711 (P. viridiflava mviN pv ).
Results and Discussion
Plasmids pIviGK and pIviGG
The genetic system that utilizes our pIviGG and pIviGK plasmids is a homologue of IVET [12], however, it has some differences. First, the application of our plasmid constructions does not require any auxotroph mutation of the bacteria under study. Additionally, the bacteriolytic activity of the kanamycin and gentamicin antibiotics used [13] provides a stringent in vivo selection pressure. As a second difference, the atpE [23] and TIS (Fig. 1) translational enhancers in the 5′ untranslated region of the reporter genes provide the monocopy application of these genes. A third difference is that the PacI recognition sequences make the multiple cloning site (MCS, Fig. 1) easily variable. An additional advantage of these new plasmids is the use of three-way translational stop codons in front of the reporter genes (Fig. 1.), that precludes the generation of non-functional fusion reporter proteins.
Characterisation of P. viridiflava ivi-genes
In the present study, we have tested pIviGK in conjunction with Pseudomonas viridiflava, proving the applicability of the created genetic constructs. It has enabled the isolation of five unique open reading frames (ORFs) from ten fusion joint points that were sequenced from 132 in vivo kanamycin-selected colonies. BlastP [1] analysis was done with putative products from translation of ORFs in the same orientation as the reporter genes. The search for protein homology has shown that IviPV-s1/7 (henceforth s1/7), contains an ORF encoding the pectate lyase of P. viridiflava [10] that attacks pectin, the major constituent of plant cell wall in the soft rot disease of fruits and vegetables. Another strain identified by IVET, IviPV-s1/6 (henceforth s1/6), contains an ORF that encodes a protein sharing 100% aa. identity with the MviN membrane protein from Pseudomonas syringae pv. tomato DC3000. This protein was first described as a virulence determinant of Salmonella typhymurium [4]. However, the biochemical function of the product encoded by mviN and similar genes is not fully understood [9]. Other ORFs that were identified as homologues to Pseudomonas syringae pv. tomato DC3000 genes may play some marginal role in the pathogenesis of P. viridiflava. These include a gene encoding deoxyguanosine-triphosphate triphosphohy- drolase (dGTPase; EC 3.1.5.1), an ABC transporter (ATP-binding/permease protein), and a K+-dependent Na+/Ca2+ exchange related-protein that matches the TIGR protein family HMM (TIGR00846).
Phenotypic analysis of MviNpv
The knockout mutant derivative of P. viridiflava, designated PV-dm, grew normally in Luria broth medium and showed no unusual colony morphology on LB agar plates. A mutation in mviN influences motility and virulence in S. typhimurium [4]. Therefore, the PV-dm mutant was tested for motility by a soft agar motility assay, which showed that a non-motile phenotype occurred through the disruption of the mviN pv gene (data not shown). Effect of the mviN pv mutation on the virulence of P. viridiflava in Arabidopsis thaliana was assayed by the growth curve analysis of the wild type and PV-dm mutant P. viridiflava and P. fluorescens. The latter strain is a non-pathogenic saprophytic bacterium that cannot multiply within Arabidopsis leaves due to its inability to invade the plant. The dipping inoculation method was used in order to model the bacterial invasion that occurs in nature. The growth curves of PV-dm differed from that of the wild type strain but were similar to that of P. fluorescens. (Fig. 2). This result suggests that the mviN pv gene is a virulence factor of P. viridiflava that contributes to invasion of plant leaves, but not to the colonisation of the leave surface before invasion. However, when Arabidopsis leaves were syringe-infiltrated with bacterial suspensions of wild type P. viridiflava and the PV-dm strain in the range of 106–109 cfu/ ml, we could not detect any differences either in their in planta growth curve (Fig. 2) or in the macroscopical appearance of provoked symptoms and the time required for symptom development (data not shown). Thus, MviNpv functions at an early stage of infection. This result corresponds to the earlier studies that have shown that motility is a common strategy employed by plant pathogenic bacteria and is part of the virulence machinery of the pathogen [5, 8, 16]. Both motile and non-motile P. syringae pv. glycinea strains are equally pathogenic when inoculated into soybean leaves [8], suggesting that motility provides an invasion advantage but is less important for multiplication once the bacteria are inside plant tissues.

Growth curves of wild type (open and filled circles) and PV-dm mutant (open and filled squares) P. viridiflava and P. fluorescens (triangles) in Arabidopsis thaliana leaves. Solid line: the infection was carried out by a dipping inoculation method, using bacterial suspensions of 108 cfu/mL. Dotted line: the inoculation was carried out by a blunt end syringe, using bacterial suspensions of 106–109 cfu/mL. Although actual inoculum densities differed between experiments, growth patterns were similar. Bacteria were enumerated in the samples in triplicates.
Activity of isolated promoters
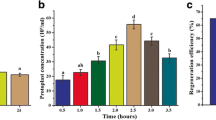
During our investi- gations, it became obvious that the gfp reporter gene makes pIviGK useful for the selection of promoters with moderate activity that can increase under inducing circumstances. Strain IviPV-s1/6 accumulated enough GFP for visual scoring only three days post infection. This suggests that the promoter region of mviN pv has a moderate basic activity that is capable of switching to an elevated expression under inducing circumstances. This hypothesis was confirmed by spectrofluorometric measurements wherein in planta activation of the mviN pv and pel genes have been examined. Figure 3 shows that in the case of mviN pv , detectable bright green light could be measured immediately after infection and 20% increase of the mean intensity was detected at 1.5 hours after infection that turned to twofold at the third hour of the measurement. In contrast, the emitted fluorescence of strain s1/7 carrying the pel-gfp fusion was above the detection baseline only at 1.5 hours post infection.

In planta expression dynamics of mviN pv and pel as measured by expression of the fused gfp reporter gene. Strains IviPV-s1/6 and IviPV-s1/7 represent mviN pv -gfp and pel-gfp fusions, respectively. Due to the fluorescent background of pepper tissue excited with 395 nm UV light, we could measure GFP emission levels only above 6.5 × 105 photon counts/sec. Thus, fluorescence intensities that correctly reported promoter activities were above 7 × 105 photon counts/sec; this value also determined the baseline of the experiment. The useful range of bacterial cells that were vacuum infiltrated into a single pepper ring for fluorescence measuring was between 107–108 cfu/sample. GFP emission of pulped pepper ring samples containing strains IviPV-s1/6 and Ivi-s1/5 of P. viridiflava were examined at 0, 1.5, 2, and 3 hours post-infection. The experiment was repeated three times and each sample was measured in triplicates.
In summary, the pIviGK plasmid construct proved to be a feasible tool for the identification of genes that are induced during the infection of plants. We have identified in planta inducible genes of P. viridiflava, which include mviN pv encoding a membrane protein and pel that produces pectate lyase, a key virulence factor of soft rot pathogens. Based on these results, the IVET method that utilizes pIviGK would certainly allow identification of additional virulence genes of plant pathogenic bacteria thereby providing a deeper insight into the pathogenesis of bacterial diseases of plants.
Literature Cited
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Angelichio MJ, Camilli A (2002) In vivo expression technology. Infect Immun 70:6518–6523
Brown DG, Allen C (2004) Ralstonia solanacearum genes induced during growth in tomato: an inside view of bacterial wilt. Mol Microbiol 53:1641–1660
Carsiotis M, Stocker BA, Weinstein DL, O’Brien AD (1989) A Salmonella typhimurium virulence gene linked to flg. Infect Immun 57:3267–3280
Cuppels D.A. (1988) Chemotaxis by Pseudomonas syringae pv. tomato. Appl Environ Microbiol 54:629–632
Figurski D. Helinski DR (1979) Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc Natl Acad Sci USA 76:1648–1652
Fumiaki K, Roger T, He SY The Arabidopsis Thaliana-Pseudomonas Syringae Interaction. In: Somerville CR, Meyerowitz EM (eds) The Arabidopsis book. Rockville, MD: American Society of Plant Biologists. http://www.aspb.org/publications/arabidopsis/
Hattermann DR, Ries SM (1989) Motility of Pseudomonas syringae pv. glycinea and its role in infection. Ecol. Epidemiol. 79:284–289
Hvorup RN, Winnen B, Chang AB, Xiao-Feng ZYJ, Saier Jr MH (2003) The multidrug/oligosaccharidyl-lipid/polysaccharide (MOP) exporter superfamily. Eur J Biochem 270:799–813
Liao C-H, Hung H-Y, Chatterjee AK (1988) An extracellular pectate lyase is the pathogenicity factor of the soft-rotting bacterium Pseudomonas viridiflava. Mol Plant-Microbe Interact 1:199–206
De Lorenzo V, Herrero M, Jakubzik U, Timmis KN (1990) Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in gram-negative eubacteria. J Bacteriol 172:6568–6572
Mahan MJ, Slauch JM, Mekalanos JJ (1993) Selection of bacterial virulence genes, that are specifically induced in host tissues. Science 259:686–688
Mardones G, Venegas A (2000) Chromogenic plate assay distinguishing bacteriolytic from bacteriostatic activity of an antibiotic agent. J Microbiol Methods 40:199–206
Olins PO, Rangwala SH (1989) A novel sequence element derived from bacteriophage T7 mRNA acts as an enhancer of translation of the lacZ gene in Escherichia coli. J Biol Chem 264:16973–16976
Osbourn AE, Barber CE, Daniels MJ (1987) Identification of plant-induced genes of the bacterial pathogen Xanthomonas campestris pathovar campestris using a promoter-probe plasmid. EMBO J 6:23–28
Panopoulos NJ, Schroth MN (1974) Role of flagellar motility in the invasion of bean leaves by Pseudomonas phaseolicola. Phytopathology 64:1389–1397
Pitcher DG, Saunders NA, Owen RJ (1989) Rapid extraction of bacterial genomic DNA with guanidium thiocyanate. Lett Appl Microbiol 8:151–156
Preston GM, Bertrand N, Rainey PB (2001) Type III secretion in plant growth-promoting Pseudomonas fluorescens SBW25. Mol Microbiol 41:999–1014
Quandt J, Hynes MF (1993) Versatile suicide vectors which allow direct selection for gene replacement in Gram-negative bacteria. Gene 127:15–21
Rainey PB, Heithoff DM, Mahan MJ (1997) Single-step conjugative cloning of bacterial gene fusions involved in microbe-host interactions. Mol Gen Genet 256:84–87
Rediers H, Bonnecarrére V, Rainey PB, Hamonts K, Vanderleyden J, DeMot R (2003) Development and application of a dapB-based in vivo expression technology system to study colonization of rice by the endophytic nitrogen-fixing bacterium Pseudomonas stutzeri A15. Appl Environ Microbiol 69:6864–6874
Sambrook J, Fritsch EF, Maniatis T (1998) Molecular cloning: A laboratory manual, 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. pp 1.25–1.28
Schauder B, McCarthy JE. (1989) The role of bases upstream of the Shine-Dalgarno region and in the coding sequence in the control of gene expression in Escherichia coli: translation and stability of mRNAs in vivo. Gene 78:59–7234
Silby MW, Levy SB (2004) Use of invivo expression technology to identify genes important in growth and survival of Pseudomonas fluorescens Pf0-1 in soil: Discovery of expressed sequences with novel genetic organization. J Bacteriol 186:7411–7419
Suarez A, Guttler A, Stratz M, Staendner LH, Timmis KN, Guzman CA (1997) Green fluorescent protein-based reporter systems for genetic analysis of bacteria including monocopy applications. Gene 196:69–74
Thanaraj TA, Pandit MW (1989) An additional ribosome-binding site on mRNA of highly expressed genes and a bifunctional site on the colicin fragment of 16S rRNA from Escherichia coli: important determinants of the efficiency of translation-initiation. Nucleic Acids Res 17:2973–2985
Acknowledgments
We are grateful to S. Lory and L. Hornok for their helpful hints and help in editing, and to J.J. Mahan, C.A. Guzmán, and A. Holczinger for providing some plasmid constructions. This research was funded by grants of the Hungarian Scientific Research Fund (OTKA T029119 and TS040835).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Czelleng, A., Bozso, Z., Ott, P. et al. Identification of Virulence-Associated Genes of Pseudomonas viridiflava Activated During Infection by Use of a Novel IVET Promoter Probing Plasmid. Curr Microbiol 52, 282–286 (2006). https://doi.org/10.1007/s00284-005-0208-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-005-0208-6