
Abstract
The predictive value of clinical and platelet kinetic parameters for treatment outcome in idiopathic thrombocytopenic purpura (ITP) was investigated in 75 patients with platelets ≤20 × 109/L. The platelet kinetic studies showed that the platelet production rate (PPR) was decreased (<100 × 109/day), normal, or increased (>355 × 109/day) in 33%, 48%, and 19% of patients, respectively. All patients started with prednisone at diagnosis (1 mg/kg/day). Initial complete and partial response (CR/PR) rate was 84% and a durable CR/PR (≥6 months without treatment) was attained in 44% of the patients. Durable CR/PR was noticed in 64% of the patients with decreased PPR during a median follow-up time without treatment of 81 (range 18–92) months, compared to 34% of the patients with normal or increased PPR during a median follow-up time without treatment of 141 (range 10–284) months (p = 0.03). Splenectomy was performed in 32% of patients with decreased PPR and in 62% of patients with normal or increased PPR (p = 0.03). In conclusion, ITP patients with suppressed PPR have a significant higher durable CR/PR rate to prednisone therapy and are less frequently exposed to splenectomy than those with a normal or increased PPR.
Similar content being viewed by others


Introduction
Thrombocytopenia in idiopathic thrombocytopenic purpura (ITP) is predominantly caused by autoantibodies that recognize antigens on platelets and megakaryocytes resulting in accelerated platelet destruction and decreased platelet production rate (PPR) [1, 2]. Premature cell death of megakaryocytes plays an important role in the reduced platelet production [3]. However, easily applicable tests that can make a distinction between a reduced platelet half life and a reduced PPR are lacking. Circulating thrombopoietin distinguishes between disorders of platelet destruction and production [4] but has no discrimitive value within a group of ITP patients. Platelet glycocalicin, the soluble outer fragment of the α-chain of platelet glycoprotein Ib, may distinguish between thrombocytopenia due to reduced production or increased destruction [5], but its diagnostic value in patients with ITP is not well defined. In the absence of simple tests, platelet kinetic studies are still the only method to determine the relative contribution of increased peripheral platelet destruction and decreased production [6]. Many studies have been performed to analyze the predictive value of thrombokinetic parameters for the response to treatment in ITP. Most of these studies have focused on splenectomy. Although individual studies have reported a predictive value for the site of platelet sequestration for the response to splenectomy, a recent systemic review did not support this notion [7]. The predictive value of the PPR has not been extensively investigated. Siegel et al. [8] showed that an increased PPR in conjunction with a short platelet survival correlated with a favorable response to splenectomy, while patients with a reduced PPR and a moderately reduced survival demonstrated a poor outcome following splenectomy. In keeping with these findings are platelet kinetic studies using radioactive indium (111In), which have shown that splenectomy improves the platelet counts predominantly by increasing platelet survival [9, 10]. In these studies [9, 10], prednisone primarily affected the platelet production, which contradicted earlier radiolabeled platelet studies using chromium-51 (51Cr), suggesting that successful response to prednisone was caused by an increase in platelet survival [11]. In the present study, we investigated the predictive value of thrombokinetic and clinical parameters in 75 adult patients with severe ITP. The results demonstrate that patients with a decreased PPR have a significantly higher durable complete and partial response (CR/PR) to prednisone, and subsequently are less frequently exposed to splenectomy than patients with a normal or increased PPR.
Materials and methods
Patients
Seventy-five untreated adult patients with severe ITP were investigated after informed consent was obtained. The institutional review board of the University Hospital Groningen approved the study protocol. The diagnosis was made according to the guidelines on ITP from the American Society of Hematology [12]. This requires that thrombocytopenia in an otherwise healthy person remains of undetermined etiology after history, physical examination, complete blood count, and examination of the peripheral blood smear. Patients with associated systemic diseases, including human immunodeficiency virus infection, were excluded. Consecutive patients with a platelet count ≤20 × 109/L were enrolled from 1 January 1977 until 1 November 2005. Patients who were receiving treatment for ITP, including glucocorticoids and intravenous immunoglobulins, at the time of radiolabeled studies and patients who underwent platelet kinetic studies during the course of the disease or at the time of relapse were also not included. Patients with a complete or partial response (PR) to prednisone needed to be at least 6 months without prednisone or other treatment for ITP at the moment of evaluation.
Treatment strategy and definitions
Treatment with prednisone (1 mg kg−1 day−1) was started at a platelet count ≤20 × 109/L irrespective of the presence of bleeding. The protocol prescribed that splenectomy should be performed if there was no initial response to prednisone after 4–6 weeks of therapy, when relapse of thrombocytopenia occurred after tapering prednisone, or in the case of a severe hemorrhagic diathesis.
A patient was considered to have a complete response (CR) if the platelet count increased to ≥150 × 109/L, and a PR when the platelet count varied between 30 to 150 × 109/L. No response (NR) was defined if the platelet count remained unchanged or increased to less than 30 × 109/L. Additionally, a distinction between initial and durable CR and PR was made. A CR or PR was considered durable if it persisted for more than 6 months after cessation of therapy. Initial PR or CR to prednisone or splenectomy was defined as rise in the platelet count ≥30 and ≥150 × 109/L, respectively, within 3 months after initiating therapy. Patients who had an initial response to prednisone but relapsed during tapering of prednisone were considered as nonresponders. The rate and duration of tapering was left to the discretion of the treating physician.
The observation period (defined as follow-up time) started on the day of initial diagnosis and finished on January 1, 2006, or when a patient died (due to any cause). Long-term clinical follow-up data were obtained in 72 patients. Three patients were lost during follow up. The relapse-free follow-up time after a certain treatment is defined as months between the date of reaching initial CR or PR to this treatment and the date of relapse defined by platelets ≤30 × 109/L, splenectomy, death, or the final date of the study. Time to splenectomy indicates months between the date of diagnosis and the date of splenectomy. Time to splenectomy failure is defined as months between the date of splenectomy and a decrease in the platelet count to ≤30 × 109/L.
Platelet kinetic studies
111In-tropolonate was used for labeling [concentration 25 μg/mL, activity 37 MBq Indium-111 at calibration time (Mallinckrodt Medical, Petten, The Netherlands, BV)]. Intravenous blood was collected in polypropylene syringes (Becton and Dickinson, Mountain View, CA, USA), and the labeling procedure was performed in polypropylene tubes (Falcon 2070, Becton and Dickinson). Acid citrate dextrose was used as anticoagulant, according to the recommendations of the National Institute of Health (22 g trisodium citrate, 8 g citric acid, and 25 g dextrose per liter).
Labeling was performed following the recommendations of the International Committee on Standardization in Hematology [13]. Briefly, autologous blood was collected in acid-citrate-dextrose solution and centrifuged at 200 g. Platelet-rich plasma was harvested and acidified (pH 6.5) and the platelets were pelleted by centrifugation. The pellet was resuspended in autologous platelet-poor plasma and labeled with 10 MBq 111In-tropolonate. After labeling efficiency was established (usually >90%), a dose of approximately 7 MBq radiolabeled platelets was injected. Before injection, the platelet suspension was evaluated for lymphocyte and red cell contamination.
Initial platelet recovery, mean platelet lifespan, and PPR were calculated in whole blood samples taken at intervals of 1, 3, and 5 h after injection and twice a day for 4 days thereafter. Additional blood samples were drawn on the first day if reduced mean platelet lifespan was expected. Extrapolation of the platelet survival curve to time zero was used in the determination of platelet recovery. The initial platelet recovery is defined as the percentage of platelets circulating in the blood immediately after injection. Mean platelet lifespan was calculated using the multiple hit model, following the recommendations of the International Committee of Standardization in Hematology [13]. Blood volume in liters was estimated from height and weight [14]. PPR was calculated from mean platelet lifespan, platelet count, and initial platelet recovery following the description of Harker and Finch [15] and multiplied by the blood volume, and therefore expressed as platelets/day. At stable platelet counts, platelet turnover (i.e., the rate of platelet destruction) equals PPR. The PPR can therefore be defined as the number of platelets entering the circulation to maintain the platelet count. This is not necessarily identical to the real platelet production because production in the bone marrow without release of platelets into the circulation may occur. Normal values, expressed as median (range), are a mean platelet lifespan of 9.2 (7.4–11) days, a PPR of 223 (100–355) × 109/day, and an initial platelet recovery of 60 (45–88) percent. These values were obtained from platelet kinetic studies in 11 healthy volunteers, who were recruited from laboratory personnel and hospital staff [16]. PPR was defined as decreased when <100 × 109/day, and increased when >355 × 109/day.
Organ uptake measurements
Platelet destruction took place in the spleen or liver or both, or diffusely in the reticuloendothelial system. Hepatic and splenic uptakes were measured with a large-field-of-view gamma camera interfaced with a computer (ICON, Siemens, Chicago, IL, USA). Immediately after injection of the labeled platelets, 60 1-min frames were recorded in the posterior position. Dynamic investigation was followed by 5-min static acquisition in both anterior and posterior positions. Twice a day, these views were repeated and blood samples were taken to calculate absolute uptake, platelet activity in liver and spleen [17], and the geometric mean of the liver and spleen.
In normal subjects, hepatic and splenic uptake remains relatively constant during the 5 days. Thus, apart from the pooling phenomenon in the first hour after injection, increased splenic or hepatic uptake during the next few days indicates increased platelet destruction. In patients with nonsplenic sequestration, platelets are destroyed in the liver or diffusely in the reticuloendothelial system.
Statistical analysis
Continuous data are reported as median (range). Statistical comparisons were performed using the Mann Whitney U test, Chi-square, and Fisher’s exact test. Correlation was assessed with the Spearman’s rank correlation procedure. The Kaplan–Meier method was used to calculate relapse-free survival (RFS) curves. Statistical significance of differences between the groups was assessed using the log-rank test. Findings were considered statistically significant at the p < 0.05 level, and all tests were two-sided.
Results
Seventy-five patients with severe ITP (platelet count ≤20 × 109/L) were evaluated. Patient characteristics are shown in Table 1. Platelet kinetic studies were performed in all studied patients. Total follow-up time after diagnosis was a median of 132 (13–348) months. Seventy (93%) patients had a follow up of more than 3 years. Sixteen patients (21%) died during follow up at a median of 81 (13–293) months after diagnosis. Median age at the time of death was 81 (33–95) years. Fourteen patients died of causes unrelated to ITP or treatment for ITP. The causes were mainly cardiopulmonary-related in 12 patients, while two patients (at age of 39 and 40 years) died of disseminated malignancy that developed years after the diagnosis of ITP was established. Of these 14 patients, nine patients had reached a durable CR/PR to prednisone, two patients had a durable CR to splenectomy and two patients were refractory to prednisone and splenectomy but reached a CR to an alternative therapy, and one patient had NR to prednisone and splenectomy and was not treated afterwards. Two patients died at the ages of 33 and 70 years of uncontrolled bleeding during severe thrombocytopenia due to refractory ITP. Three patients were lost to follow-up 27, 60, and 72 months after diagnosis. Of these three patients, two had a durable CR after splenectomy and one patient had NR to prednisone but had stable platelet counts ≤30 × 109/L without bleeding complications with no therapy and refused splenectomy.
Response to treatment
All 75 patients were treated with prednisone; 63 (84%) had an initial CR or PR to prednisone whereas 33 patients (44%) had a durable CR/PR to prednisone therapy. Following discontinuation of prednisone, these patients were relapse-free during a median follow-up time of 99 (10–284) months. Kaplan Meier analysis for RFS following prednisolone therapy is shown in Fig. 1. RFS estimate was 44% [95% CI (32%–56%)] and the median RFS time was 38 [95% CI (0–101)] months (mean 135 [95% CI (102–168)] months). Patients who reached a durable response to prednisone were also not treated with additional medications. Eleven patients did not respond to prednisone at all and underwent splenectomy. One additional nonresponding patient refused splenectomy and was treated for a longer period (12 months) with low-dose prednisone. Thirty patients relapsed during tapering of prednisone therapy. Two of these patients refused splenectomy and 28 patients were subsequently splenectomized. Characteristics of the patients according to durable CR/PR and NR to prednisone therapy are shown in Table 2. With regard to most characteristics, no significant differences were observed between the two groups except for the relative number of patients with a decreased PPR, which was significantly higher in the group with a durable CR/PR (p = 0.03).

RFS of ITP patients following prednisone and following splenectomy
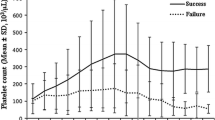
Of the patients (n = 25) with a decreased PPR (<100 × 109/day), 16 (64%) reached a durable CR/PR to prednisone, compared to 17 (34%) of the patients (n = 50) with a normal or increased PPR (≥100 × 109/day) (p = 0.03). Following discontinuation of prednisone, the patient groups were relapse-free during a median follow up of 81 (18–192) and 141 (10–284) months, respectively. Figure 2 shows Kaplan Meier estimates of RFS for patients with decreased or normal/increased PPR following prednisone therapy. For the patients with a decreased PPR, RFS estimate was 64% [95% CI (45%–85%)] and mean RFS time was 133 months [95% CI (97–170)]. For the patients with a normal or increased PPR, the RFS estimate was 34% [95% CI (20%–48%)] and median RFS time was 10 months [95% CI (0%–27%)] (mean 107 [95% CI (69–145)]). Characteristics of the patient subgroups are depicted in Table 3. Among patients with a normal or increased PPR, the mean platelet lifespan was significantly shorter and splenic sequestration was significantly more frequent.

RFS of ITP patients following prednisone therapy by level of PPR
Splenectomy
Thirty-nine (93%) of the nonresponding patients (n = 42) underwent splenectomy. Characteristics of nonresponding patients are shown in Table 2. Time to splenectomy was 5 (1–82) months. Twenty-nine (74%) patients reached a durable CR/PR to splenectomy only. Figure 1 shows the RFS of the splenectomized patients compared to the patients following prednisone therapy. RFS estimate for the subgroup that underwent splenectomy was 74% [95% CI (59%–88%)] and the mean RFS time was 191 months [95% CI (157–225)]. For the nonresponders to splenectomy, the time to splenectomy failure was 2 (0–94) months. After splenectomy, four of 39 patients (10%) obtained a CR/PR on cyclosporine, danazol or vincristin. Six (8%) of 77 patients were refractory to any treatment. There was no significant difference between responders and nonresponders to splenectomy in patient age (p = 1.0), sex (p = 0.6), platelet count at diagnosis (p = 0.2), mean platelet lifespan (p = 0.3), PPR (p = 0.4), and site of platelet sequestration (spleen, p = 0.7 and liver, p = 0.1) and time to splenectomy (p = 1.0). These parameters were also not significantly different between the groups with PPR <100 × 109/day and ≥100 × 109/day.
Discussion
In the present study, patients with severe ITP were investigated for associations between clinical characteristics, thrombokinetic parameters, and the response to treatment with prednisone or splenectomy. The results demonstrate that patients with a decreased PPR had a significantly higher durable CR or PR rate to prednisone therapy than those with a normal or increased PPR. This was also reflected in the significantly lower splenectomy rate in the group of patients with a decreased PPR.
In ITP, therapy is generally not required when platelet counts are above 20 to 30 × 109/L [12, 18]. Glucocorticoids are usually the first choice of treatment for ITP. Initial response rates of 50–90% have been reported depending on the intensity and duration of therapy, but only 20–30% of patients enter a prolonged remission, usually defined as lasting more than 6 months after cessation of therapy [18–22]. The overall long-term remission rate to prednisone in the present study (44%) is higher than these reported previously. Variations among the different studies with regard to definitions of CR/PR, patient characteristics, timing of splenectomy, and the pooling of different remission rates may be responsible for these discrepancies. Moreover, there is a large variation (3% to 50%) in reported remission rates to glucocorticoids among the different studies [12].
Several factors, including the duration of symptoms before starting glucocorticoids, the speed of response, age, sex, and initial platelet count, have been associated with a favorable outcome of glucocorticoid treatment, but the reproducibility of these associations among the different studies has been low [22–27]. None of these factors correlated with a response to prednisone in our analysis. Although there was a high death rate in our patient cohort, this was mainly due to the long follow-up time, and the demographics are not different from ITP populations studied previously.
To our knowledge, the predictive value of the PPR for prednisone therapy in ITP has not been reported so far. Previous studies have shown that prednisone improves the platelet count in ITP primarily by increasing platelet production [9, 10]. The present findings are consistent with these results by showing that ITP patients with the lowest PPR are the most likely to respond to prednisone in the long term. In addition, patients with a decreased PPR showed significantly less splenic sequestration and a significantly longer mean platelet lifespan than patients with a normal or increased PPR. These findings are consistent with previous studies [8, 28–30] and further underscore the heterogeneity of the disease. Thus, in patients with a normal or elevated PPR, increased peripheral destruction of platelets contributes relatively more to thrombocytopenia than suppressed production, which is reflected by a shorter mean platelet lifespan and a higher rate of splenic uptake.
Since an enhanced platelet production would be the normal compensatory response of the bone marrow to thrombocytopenia due to peripheral destruction, ITP patients with a normal PPR can also have an ineffective platelet production. In autoimmune hemolytic anemia, the compensatory reaction is much more pronounced, except when circulating antibodies are directed to normoblasts [31]. Previous studies have shown that, in the majority of ITP patients, the PPR is either reduced or normal [28, 30, 32–36]. Additional support for a suppressed platelet production in ITP comes from in vitro studies showing that megakaryocyte production and maturation can be inhibited by antiplatelet antibodies [1, 2, 37]. Similar in vitro cultured megakaryocytes in the presence of ITP plasma show features compatible with premature cell death [3]. The variability in PPR among different individuals is therefore likely to be related to the degree of autoantibody binding to megakaryocyte antigens. Other mechanisms, including capacity and function of the reticuloendothelial system, the degree of autoantibody-induced activation of complement, and T-lymphocyte-mediated cytotoxity may also play a role [1]. We did not observe a difference in response rates to prednisone between patients with normal and increased PPRs. This might be due to the small number of patients studied.
Different mechanisms have been proposed for explaining the beneficial effects of prednisone on the reduced platelet production, including impairment of autoantibody binding to megakaryocytes [9, 38, 39], an altered Fcγ-receptor-mediated immune clearance [40, 41], and a reduction of antibody production due to a lympholytic effect [42–44]. In addition, a reduction of premature cell death of ITP megakaryocytes has been observed during in vivo prednisone therapy [3].
That reduced platelet production is an important mechanism in ITP in vivo is also supported by recent studies with the novel thrombopoiesis-stimulating agents, such as thrombopoietin mimetic AMG-531. Short-term use of AMG-531 increased platelet counts in about 75% of ITP patients [45], whereas preliminary results show sustained platelet responses in about 50% of patients [46]. It is conceivable that these agents primarily promote the PPR without modulating the platelet half life. In summary, patients with severe ITP and a decreased PPR demonstrate an approximately two-times-higher long-term remission rate to prednisone and are significantly less likely to need splenectomy than ITP patients with a normal or increased PPR.
References
McMillan R, Wang L, Tomer A, Nichol J, Pistillo J (2004) Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood 103:1364–1369 doi:10.1182/blood-2003-08-2672
Chang M, Nakagawa PA, Williams SA, Schwartz MR, Imfeld KL, Buzby JS et al (2003) Immune thrombocytopenic purpura (ITP) plasma and purified ITP monoclonal autoantibodies inhibit megakaryocytopoiesis in vitro. Blood 102:887–895 doi:10.1182/blood-2002-05-1475
Houwerzijl EJ, Blom NR, van der Want JJ, Esselink MT, Koornstra JJ, Smit JW et al (2004) Ultrastructural study shows morphologic features of apoptosis and para-apoptosis in megakaryocytes from patients with idiopathic thrombocytopenic purpura. Blood 103:500–506 doi:10.1182/blood-2003-01-0275
Emmons RV, Reid DM, Cohen RL, Meng G, Young NS, Dunbar CE et al (1996) Human thrombopoietin (TPO) levels are high when thrombocytopenia is due to megakaryocyte deficiency and low when due to increased platelet destruction. Blood 87:4068–4071
Steinberg MH, Kelton JG, Coller BS (1987) Plasma glycocalicin. An aid in the classification of thrombocytopenic disorders. N Engl J Med 317:1037–1042
Hersh J (2000) Mathematical analysis of the relative contributions of decreased production and increased peripheral destruction in idiopathic thrombocytopenic purpura and implications in splenectomy. J Theor Biol 203:153–162 doi:10.1006/jtbi.2000.1072
Kojouri K, Vesely SK, Terrell DR, George JN (2004) Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 104:2623–2634 doi:10.1182/blood-2004-03-1168
Siegel RS, Rae JL, Barth S, Coleman RE, Reba RC, Kurlander R et al (1989) Platelet survival and turnover: important factors in predicting response to splenectomy in immune thrombocytopenic purpura. Am J Hematol 30:206–212 doi:10.1002/ajh.2830300404
Gernsheimer T, Stratton J, Ballem PJ, Slichter SJ (1989) Mechanisms of response to treatment in autoimmune thrombocytopenic purpura. N Engl J Med 320:974–980
Louwes H, Vellenga E, Houwerzijl EJ, de Wolf JT (2001) Effects of prednisone and splenectomy in patients with idiopathic thrombocytopenic purpura: only splenectomy induces a complete remission. Ann Hematol 80:728–732 doi:10.1007/s002770100375
Branehög I, Weinfeld A (1974) Platelet survival and platelet production in idiopathic thrombocytopenic purpura (ITP) before and during treatment with corticosteroids. Scand J Haematol 12:69–79
George JN, Woolf SH, Raskob GE, Wasser JS, Aledort LM, Ballem PJ et al (1996) Idiopathic thrombocytopenic purpura: a practice guideline developed by explicit methods for the American Society of Hematology. Blood 88:3–40
ICSH Panel on Diagnostic Application of Radionuclides (1988) Recommended method for Indium-111 platelet survival studies. J Nucl Med 29:564–566
Bowring CS (1981) Radionuclide tracer techniques in haematology. Butterworths, London, pp 40–41
Harker LA, Finch CA (1969) Thrombokinetics in man. J Clin Invest 48:963–974 doi:10.1172/JCI106077
Louwes H, van Schuur JJ (1990) Platelet labelling with 111In-tropolonate. In: Kessler CH, Hardeman MR, Henningsen H, Petrovici JN (eds) Clinical application of radiolabelled platelets. Kluwer Academic, Dordrecht, pp 45–60
Fleming JS (1979) A technique for the measurement of activity using a gamma camera and computer. Phys Med Biol 24:176–180 doi:10.1088/0031-9155/24/1/017
Cines DB, McMillan R (2005) Management of adult idiopathic thrombocytopenic purpura. Annu Rev Med 56:425–442 doi:10.1146/annurev.med.56.082103.104644
Thompson RL, Moore RA, Hess CE, Wheby MS, Leavell BS (1972) Idiopathic thrombocytopenic purpura. Long-term results of treatment and the prognostic significance of response to corticosteroids. Arch Intern Med 130:730–734 doi:10.1001/archinte.130.5.730
Cortelazzo S, Finazzi G, Buelli M, Molteni A, Viero P, Barbui T (1991) High risk of severe bleeding in aged patients with chronic idiopathic thrombocytopenic purpura. Blood 77:31–33
Pizzuto J, Ambriz R (1984) Therapeutic experience on 934 adults with idiopathic thrombocytopenic purpura: Multicentric trial of the Cooperative Latin American Group on Hemostasis and Thrombosis. Blood 64:1179–1183
Ikkala E, Kivilaakso E, Kotilainen M, Hastbacka J (1978) Treatment of idiopathic thrombocytopenic purpura in adults. Long-term results in a series of 41 patients. Ann Clin Res 10:83–86
Meyers MC (1961) Results of treatment in 71 patients with idiopathic thrombocytopenic purpura. Am J Med Sci 242:295–302 doi:10.1097/00000441-196109000-00004
DiFino SM, Lachant NA, Kirshner JJ, Gottlieb AJ (1980) Adult idiopathic thrombocytopenic purpura. Clinical findings and response to therapy. Am J Med 69:430–442 doi:10.1016/0002-9343(80)90016-9
den Ottolander GJ, Gratama JW, de Koning J, Brand A (1984) Long-term follow-up study of 168 patients with immune thrombocytopenia. Implications for therapy. Scand J Haematol 32:101–110
Jacobs P, Wood L, Dent DM (1986) Results of treatment in immune thrombocytopenia. Q J Med 58:153–165
Stasi R, Stipa E, Masi M, Cecconi M, Scimo MT, Oliva F et al (1995) Long-term observation of 208 adults with chronic idiopathic thrombocytopenic purpura. Am J Med 98:436–442 doi:10.1016/S0002-9343(99)80342-8
Grossi A, Vannucchi AM, Casprini P, Guidi S, Rafanelli D, Pecchioli MG et al (1983) Different patterns of platelet turnover in chronic idiopathic thrombocytopenic purpura. Scand J Haematol 31:206–214
Stratton JR, Ballem PJ, Gernsheimer T, Cerqueira M, Slichter SJ (1989) Platelet destruction in autoimmune thrombocytopenic purpura: kinetics and clearance of indium-111-labeled autologous platelets. J Nucl Med 30:629–637
Louwes H, Zeinali Lathori OA, Vellenga E, de Wolf JT (1999) Platelet kinetic studies in patients with idiopathic thrombocytopenic purpura. Am J Med 106:430–434 doi"10.1016/S0002-9343(99)00054-6
Van De Loosdrecht AA, Hendriks DW, Blom NR, Smit JW, De Wolf JT, Vellenga E (2000) Excessive apoptosis of bone marrow erythroblasts in a patient with autoimmune haemolytic anaemia with reticulocytopenia. Br J Haematol 108:313–315 doi:10.1046/j.1365-2141.2000.01867.x
Heyns AP, Lotter MG, Badenhorst PN, de Kock F, Pieters H, Herbst C et al (1982) Kinetics and sites of destruction of 111Indium-oxine-labeled platelets in idiopathic thrombocytopenic purpura: a quantitative study. Am J Hematol 12:167–177 doi:10.1002/ajh.2830120209
Stoll D, Cines DB, Aster RH, Murphy S (1985) Platelet kinetics in patients with idiopathic thrombocytopenic purpura and moderate thrombocytopenia. Blood 65:584–588
Schmidt KG, Rasmussen JW (1985) Kinetics and distribution in vivo of 111In-labelled autologous platelets in idiopathic thrombocytopenic purpura. Scand J Haematol 34:47–56
Heyns AP, Badenhorst PN, Lotter MG, Pieters H, Wessels P, Kotze HF (1986) Platelet turnover and kinetics in immune thrombocytopenic purpura: results with autologous 111In-labeled platelets and homologous 51Cr-labeled platelets differ. Blood 67:86–92
Ballem PJ, Segal GM, Stratton JR, Gernsheimer T, Adamson JW, Slichter SJ (1987) Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest 80:33–40 doi:10.1172/JCI113060
Rolovic Z, Baldini M, Dameshek W (1970) Megakaryocytopoiesis in experimentally induced immune thrombocytopenia. Blood 35:173–188
Dixon R, Rosse W, Ebbert L (1975) Quantitative determination of antibody in idiopathic thrombocytopenic purpura. Correlation of serum and platelet-bound antibody with clinical response. N Engl J Med 292:230–236
Tate DY, Sorenson RL, Gerrard JM, White JG, Krivit W (1977) An immunoenzyme histochemical technique for the detection of platelet antibodies from the serum of patients with idiopathic (autoimmune) thrombocytopenic purpura (ITP). Br J Haematol 37:265–275 doi:10.1111/j.1365-2141.1977.tb06843.x
Shulman NR, Weinrach RS, Libre EP, Andrews HL (1965) The role of the reticuloendothelial system in the pathogenesis of idiopathic thrombocytopenic purpura. Trans Assoc Am Physicians 78:374–390
Handin RI, Stossel TP (1978) Effect of corticosteroid therapy on the phagocytosis of antibody-coated platelets by human leukocytes. Blood 51:771–779
Fujisawa K, Tani P, Piro L, McMillan R (1993) The effect of therapy on platelet-associated autoantibody in chronic immune thrombocytopenic purpura. Blood 81:2872–2877
Berchtold P, Wenger M (1993) Autoantibodies against platelet glycoproteins in autoimmune thrombocytopenic purpura: their clinical significance and response to treatment. Blood 81:1246–1250
McMillan R, Longmire RL, Tavassoli M, Armstrong S, Yelenosky R (1974) In vitro platelet phagocytosis by splenic leukocytes in idiopathic thrombocytopenic purpura. N Engl J Med 290:249–251
Bussel JB, Kuter DJ, George JN, McMillan R, Aledort LM, Conklin GT et al (2006) AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med 355:1672–1681 doi:10.1056/NEJMoa054626
Kuter DJ, Bussel JB, Lyons RM, Pullarkat V, Gernsheimer TB, Senecal FM et al (2008) Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 371:395–403
Acknowledgements
We wish to thank the internists from the Martini Ziekenhuis in Groningen, the Wilhelmina Ziekenhuis in Assen, the Sint Lucas Ziekenhuis in Winschoten, the Refaja Ziekenhuis in Stadskanaal, the St. Antonius Ziekenhuis in Sneek, and Delfzicht Ziekenhuis in Delfzijl for sending their patients. Ms. S. Tapken and Mrs. V. Sebo are kindly acknowledged for measuring plasma glycocalicin.
Authorship Contribution:
E.J.H designed experiments, enrolled patients, analyzed data, and wrote the manuscript. H.L. performed experiments and analyzed data. W.J.S. analyzed data and revised the manuscript. J.W.S. designed research, analyzed data, and revised the manuscript. E.V. designed research, analyzed data, and revised the manuscript. J.Th.M.d.W. designed research, analyzed data, and revised the manuscript.
Conflict of interest disclosure
The authors declare no competing financial interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Financial support was provided by a grant from the J.K. de Cock Stichting.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Houwerzijl, E.J., Louwes, H., Sluiter, W.J. et al. Platelet production rate predicts the response to prednisone therapy in patients with idiopathic thrombocytopenic purpura. Ann Hematol 87, 975–983 (2008). https://doi.org/10.1007/s00277-008-0537-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-008-0537-1