
Abstract
The MITO-END3 trial compared carboplatin and paclitaxel (CP) with avelumab plus carboplatin and paclitaxel (CPA) as first-line treatment in endometrial cancer (EC) patients and demonstrated a significant interaction between avelumab response and mismatch repair status. To investigate prognostic/predictive biomarker, 29 MITO-END3-EC patients were evaluated at pretreatment (B1) and at the end of CP/CPA treatment (B2) for peripheral myeloid-derived suppressor cells (MDSC) and Tregs. At B2, effector Tregs frequency was significantly higher in patients treated with CPA as compared to CP (p = 0.038). Both treatments (CP/CPA) induced significant decrease in peripheral M-MDSC (− 5.41%) in TCGA 2-MSI-high as compared to TCGA-category 4 tumors (p = 0.004). In accordance, both treatments induced M-MDSCs (+ 5.34%) in MSS patients as compared to MSI-high patients (p = 0.001). Moreover, in a subgroup of patients, primary tumors were highly infiltrated by M-MDSCs in MSS as compared to MSI-high ECs. A post hoc analysis displayed higher frequency of M-MDSCs (p = 0.020) and lower frequency of CD4+ (p < 0.005) at pretreatment in EC patients as compared to healthy donors. In conclusion, the peripheral evaluation of MDSCs and Tregs correlated with molecular features in EC treated with CP/CPA and may add insights in identifying EC patients responder to first-line chemo/chemo-immunotherapy.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Endometrial cancer (EC) is the most frequently diagnosed gynecologic cancer (GC) [1] with peculiar increasing in incidence and mortality [2, 3]. While the prognosis is favorable for early stage, advanced disease displays 5-year survival of 15% (13.2–17.3%) [1]. EC first-line recurrent/advanced standard treatment recommends carboplatin/paclitaxel or hormonal therapy based on histological and clinical features [4, 5]. Nearly 50% of patients progress within a year [6] and there is no efficient second-line chemotherapy or hormone therapy [7]. Recently, four TCGA molecular categories were identified: (1) DNA polymerase epsilon (POLE)/ultramutated, (2) microsatellite unstable (MSI)/hypermutated, (3) low degree of somatic copy-number alteration (SCNA), and (4) high degree SCNA [8, 9] with a gain of ≥ 5 copies or homozygous deletion (< 10 Mb) [10]. POLE/ultramutated and MSI/hypermutated tumors make ECs a reasonable candidate for immunotherapy [11]. The TCGA molecular risk classifier [12, 13] identifies groups with divergent prognoses [14], with (i) POLE mutated and mismatch repair deficient (MMRd), uncommon and with excellent prognosis; (ii) the p53 abnormal group with the poorest prognosis; (iii) no specific molecular profile (NSMP), namely, no MSI or POLE mutations and low SCNA/no p53 mutations, largest group [15], lackings univocal prognostic significance [16, 17]. Several clinical trials evaluated the addition of immunocheck point inhibitors (ICIs) to standard first-line chemotherapy in advanced/recurrent EC [18]. NRG-GY018 study enrolled 816 patients to receive pembrolizumab with paclitaxel plus carboplatin. Consistent reduction in risk of progression (PFS) or death (OS) was detected in pembrolizumab treated, deficient mismatch repair patients (dMMR) (70%) and in the proficient mismatch repair patients pMMR (46%) [19]. The RUBY study evaluated the anti-PD-1, dostarlimab, as maintenance in combination with niraparib, in 607 primary advanced or recurrent ECs. A statistically significant PFS benefit was detected in treated patients with a particular benefit in dMMR subgroups [20, 21]. The AtTEnd study evaluated carboplatin, paclitaxel, and the anti-PD-L1 atezolizumab in 551 advanced or recurrent ECs [22] showing significant survival benefit in the dMMR atezolizumab treated patients [22]. The DUO-E trial [19] evaluated the anti-PD-L1 durvalumab, and olaparib in 718 advanced or recurrent ECs. Similarly to NRG-GY018, in DUO-E improvement in PFS was observed for the durvalumab arm irrespective of MMR status, although the greatest benefit was detected in the dMMR subgroups [19]. In MITO-END3 trial, anti-PD-L1, avelumab was beneficial in MSI-high but detrimental in MSS/TP53-mutated [23].
In EC, the tumor microenvironment (TME) orients toward the suppression as Tregs/CD8+ or Treg cell/CD4+ ratios were significantly higher [24]. Tumors with high mutational load engender stronger immune responses which in turn promote prolonged patient survival. POLE-Ultramutated and MMRd subtypes are enriched for tumor-infiltrating lymphocytes (TIL), although 22% of tumors belonging to these categories display TIL-low. Conversely, p53abn and p53wt are generally TIL-low tumors, yet also contained significant proportions of TIL-high tumors [25]. Additionally, T cell exhaustion markers (PD-1, CTLA4, LAG3, TIM3, TIGIT) might be expressed in “hot” tumors [26,27,28]. Thus, all molecular subtypes can present with immunologically “hot” and “cold” tumors, suggesting that the molecular subtype may not be exhaustive to stratify patients for immunotherapy [25]. Myeloid-derived suppressor cells (MDSC) represent heterogeneous cell population with potent immune suppressive activity [29]. MDSC are immature or early myeloid cells (eMDSC)(Lin-HLA-DR-CD11b+CD33+), monocytic (M-MDSC) (CD11b+CD14+CD15−) and polymorphonuclear (PMN-MDSC), (CD11b+CD14−CD15+) [29, 30] that inhibits T cells activity through arginase-1 (Arg-1), nitric oxide (NO), and reactive oxygen species (ROS). In EC, monocytic-MDSC (M-MDSC) are more represented and highly suppressive. M-MDSC attracted to tumors via CCL2 rapidly differentiate to tumor associated macrophages (TAM) [31,32,33]. In ipilimumab-treated melanoma patients, M-MDSC variation correlates with overall survival [34], and in ECs, MDSCs infiltration associated with unfavorable clinic pathological parameters, bone marrow, and lymph node metastases [35, 36] as well as lymph node premetastatic niche formation [37].
Tregs, CD4+CD25+ lymphocytes [38], comprise three functionally and phenotypically distinct subpopulations: naïve Tregs (CD45RA+ FoxP3lo), activated/effector-type Tregs (CD45RA−FoxP3hi), sharing in vitro suppressive function and non-suppressive T cells, cytokine-secreting (CD45RA−FoxP3lo) (non-Tregs) [39,40,41]. In ECs, Tregs play a controversial role as FoxP3 significantly associated with poorer outcome [42] or with prognostic surrogate of good prognosis [43]. Grading, deep invasion, and FoxP3 or FoxP3/vessel density correlated with [44,45,46] prognosis and EC-MMRd exhibit a significantly higher number of FoxP3+ Treg cells [43, 47].
The recruitment of Tregs cells [48, 49], as for MDSCs from the peripheral blood, has been linked to immunotherapy failure [50, 51]. Thus, with the intent to identify prognostic/predictive biomarkers that could integrate the molecular categories, peripheral MDSC and Tregs were longitudinally evaluated on peripheral blood from a subgroup of patients enrolled in the MITO-END3 trial.
Material and methods
Study design and participants
This study investigated peripheral MDSCs and Tregs in advanced/recurrent EC patients treated with CP/CPA [52]. All participants were Caucasian woman with median age 65.4 years and interquartile range (IQR) from 59 to 71.6 years. Thirty-three MITO-END3 patients (15 received CP and 18 received CPA) were enrolled at Istituto Nazionale Tumori IRCCS “Fondazione G. Pascale” and peripheral blood was collected and analyzed from 29 patients, as single institute analysis. All patients provided informed consent before the initiation of trial procedures. Healthy donors (HD) (n = 23) with median 51 years IQR 43.2–54.7 were analyzed as easy implementable reference value of peripheral MDSC and Tregs (CE 36/22 oss n.1177).
Flow cytometry
EDTA peripheral blood (6 mL) samples were collected in K3 vacutainer tubes. Intracellular staining for FoxP3 was performed with a commercially available kit (BD Transcriptor Factor Buffer Set; BD Pharmingen; RRID:AB_2869424). Tregs were characterized as follows: Horizon-V450 anti-FoxP3 (clone 259D/C7; BD Biosciences Cat# 560044, RRID:AB_1645589), Pe anti-CD25 (clone 2A3; BD Biosciences Cat# 341011, RRID:AB_2783790), Pe-Cy7 anti-CD127 (clone HIL-7R-M21; clone HIL-7R-M21) BD Biosciences Cat# 560822, RRID:AB_2033938), and APC-Cy7 anti-CD4 (clone RPA-T4; BD Biosciences Cat# 557871, RRID:AB_396913) anti-CD45RA BB515 (clone HI100; BD Biosciences Cat# 564552, RRID:AB_2738841) (gate strategy in Supplementary Fig. 1). Tregs could be further differentiated in: activated/effector Tregs (a-Tregs) identified based on FoxP3HI CD45RA−, (CD4+CD25+CD127low FoxP3HICD45RA−); resting or naïve Tregs (n-Tregs) based on FoxP3intCD45RA+ (CD4+CD25+CD127low FoxP3intCD45RA+) and not true or non-Tregs based on FoxP3intCD45RA− (CD4+CD25+CD127low FoxP3intCD45RA−).
MDSCs subsets were characterized as follows: FITC Lineage Cocktail 1 (CD3, CD14, CD16, CD19, CD20, CD56; BD Biosciences Cat# 340546, RRID:AB_400053), PE anti-CD11b (clone Mac-1; BD Biosciences Cat# 557321, RRID:AB_396636), PER-CP anti-CD33 (clone P67.6; BD Biosciences Cat# 341650, RRID:AB_400242), Pe-Cy7 anti-HLA-DR (clone G46-6; BD Biosciences Cat# 555813, RRID:AB_396147), APC anti-CD15 (clone HI98; BD Biosciences Cat# 551376, RRID:AB_398501), and APC-Cy7 anti-CD14 (clone MφP9; (BD Biosciences Cat# 333945, RRID:AB_399960) (gate strategy in Supplementary Fig. 2). CD4-lymphocyte and Tregs were defined as percentages of whole blood, while MDSCs subsets were defined as percentages of CD45 expressing PBMCs. Samples were acquired by BD FACS ARIAIII (BD Biosciences, San Jose, CA, USA). Viability was verified with LIVE/DEAD cell stain (Invitrogen by ThermoFisher, Cat. #L34966). Data were analyzed using BD FACS Diva Software 8.0 cat. 23-14523-00 (BD Bioscience; RRID:SCR_001456) [53].
Fluorescent multiplex immunofluorescence
Primary tumors were evaluated for M-MDSC (CD11b+CD14+) infiltration by multiplex immunofluorescence (mIF). Sections were selected for tumor cellularity (> 70%), absence of large areas of central necrosis or fibrosis. Moreover, when possible, nonneoplastic tissue was included in the sample. M-MDSCs were quantified on the high-resolution and high-contrast images captured by THUNDER Technology (Leica Microsystems), as double-positive, CD14+, and CD11b+ cell membrane expression were considered monocytic (M-MDSC). The Opal mIF protocol (PerkinElmer—Akoya Biosciences Cat# NEL811001KT, RRID:AB_3665660) was applied. Formalin-fixed paraffin-embedded (FFPE) human endometrial cancer tissue blocks were cut at 5 μm. Tissue sections were submerged in citrate buffer (BOND Epitope Retrieval Solution 2, pH 8), and antigen retrieval was performed in a decloaking chamber for 20 min 95 °C (Biocare Medical). Tissue sections were then blocked for 30 min at room temperature in Serum-Free Protein Block (DAKO) and stained with Rabbit recombinant monoclonal antibody. Anti-CD11b antibody [EPR1344] ab133357 (Abcam Cat# ab133357, RRID:AB_2650514) was diluted in antibody diluent (DAKO) 2 h at 25 °C, followed by secondary: HRP-conjugated AKOYA biosciences Opal Polymer HRP Ms + Rb (Akoya Biosciences Cat# ARH1001EA, RRID:AB_2890927) and Opal™ 520 (1:250, PerkinElmer—Akoya Biosciences Cat# NEL811001KT, RRID:AB_3665660). After labeling was complete, antibodies are removed in a manner that does not disrupt the Opal fluorescence signal in a decloaking chamber for 20 min 95 °C with AR6 Buffer AKOYA biosciences (AR600250ML); a second run was conducted with Recombinant CD14 (EPR3653) Rabbit Monoclonal (Cell Marque Cat# 114R, RRID:AB_2827391) for 2 h at 25 °C, followed by secondary: HRP-conjugated IgG Opal Polymer HRP Ms + Rb (Akoya Biosciences Cat# ARH1001EA, RRID:AB_2890927). Substrate: Opal™ 570 (1:250 (PerkinElmer—Akoya Biosciences Cat# NEL811001KT, RRID:AB_3665660). Counterstain: DAPI (blue). Confocal images were acquired on an Leica THUNDER Imager Tissue microscope (RRID:SCR_026034). Multispectral images allow analysis of double-positive cells (positive cells/mm2). M-MDSCs were counted in at least three regions of interest (ROI)/slide, through peritumoral (along the stromal–tumor interface at tumor edge) to intratumoral region (cells totally surrounded the neoplastic cells). M-MDSC were manually enumerated in five consecutive, not-overlapping high-power fields (400× magnification—0.237 mm2/field) for each ROI and expressed in cell/mm2 for slide/patient. Immunostainings were evaluated by three independent observers (C.D., G.S., and C.M.) blinded to patients’ clinical characteristics.
Statistical analyses
Plasma samples were collected at two time points: before starting treatments (B1) and at the end of cycle 6 (B2). The variation in the concentration of each immune cell population between the two time points was calculated as follows: delta, Δ = B1–B2. Continuous variables were described using the median and interquartile range (IQR), while qualitative variables were presented in terms of absolute numbers and proportions. The distribution of all peripheral CD4, MDSCs, and Tregs immune populations was represented using histograms. The normality of the data was assessed using the Shapiro–Wilk test. In case of a violation of the normality assumption, nonparametric tests were employed. For continuous variables, differences between two independent groups were tested using the T test or Mann–Whitney U test, as appropriate, depending on the distribution of the data. To test the association between treatment and the variation of each cell population (Δ), a linear regression model was applied. Immune populations were stratified in four categories: POLE mutated (POLEmut) (category 1); MSI-high (category 2); MSS/TP53 wt; (category 3); MSS/TP53 mut (category 4) according Cancer Genome Atlas (TCGA) and microsatellite instability (MSI) molecular data. Two multivariable regression models were conducted with TCGA and MSI status serving as covariates. In the post hoc analysis, the Mann–Whitney U test was used to compare differences between baseline levels of peripheral cells between EC patients and healthy volunteers. Data were analyzed using R software version 4.2.2 (R Project for Statistical Computing; RRID: SCR_001905). The MITO-END3 trial was based on the hypothesis (tested with 80% power) that adding avelumab to carboplatin/paclitaxel would prolong PFS as the primary endpoint. All the planned analyses related to the translational part of MITO-END3 trial were conducted for descriptive purposes to generate hypotheses and did not include a statistical sample size calculation.
Results
Sample collection
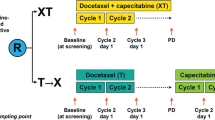
MITO-END3 is a multicenter phase 2 trial that showed improving in PFS in the dMMR population following the addition of avelumab to carboplatin and paclitaxel (CP) in first-line treatment of ECs [52]. To identify possible predictive peripheral biomarkers, a sub-cohort of 29/33 MITO-END3 patients (15 received CP and 18 received CPA) enrolled at Istituto Nazionale Tumori IRCCS “Fondazione G. Pascale” were evaluated. Baseline patient characteristics are shown in Table 1. Peripheral blood samples were collected before starting treatments (B1) and at the end of cycle 6 (B2). B1 and B2 paired samples analysis were available for 27 patients. Distribution of immune cell populations at B1 and B2 was described in Supplementary Fig. 3. Twenty-three/27 patients were characterized for TCGA and MSI status (Fig. 1). Longitudinal variations in peripheral cell population were analyzed (delta, Δ = B1–B2).

Study profile. Blood Tregs and MDSCs were evaluated at B1 (pretreatment) and at B2 (post 6 cycles of CP/ CPA). Flow cytometry identified Monocytic CD14+HLA-DR−/low-(M-MDSC), early stage Lin−HLA-DR−CD33+-(E-MDSC) and polymorphonuclear CD14−CD15+CD11b+ (PMN-MDSC); activated/effector Tregs (a-Tregs) identified based on FoxP3HI CD45RA−, (CD4+CD25+CD127low FoxP3HICD45RA−); resting or naïve Tregs (n-Tregs) based on FoxP3intCD45RA+ (CD4+CD25+CD127low FoxP3intCD45RA+) and not true or non-Tregs based on FoxP3intCD45RA− (CD4+CD25+CD127low FoxP3intCD45RA−) from MITO-END3-EC patients
M-MDSCs frequency decreases over treatment in TCGA-category 2 (MSI-high) patients in comparison with TCGA-category 4 (MSS)
ECs patients were stratified according to the TCGA classification [12, 13] (supplementary Tables 1 and 2). The association between TCGA and variations in circulating immune cell populations (Δ) was studied in 23 patients for which B1 and B2 samples were available (Table 2). Over treatment, either CP or CPA, a significant M-MDSCs decrease Δ of 5.41% (95% CI 2.34, 8.50; p = 0.004) was detected in TCGA-category 2 (MSI-high) patients in comparison with TCGA-category 4 (MSS). Thus, TCGA-category 4 ECs displayed a significant increase in M-MDSC Δ of 5.41% (95% CI 2.34, 8.50; p = 0.004) as compared to TCGA-category 2 (Table 2). Accordingly, when patients were stratified based on MSI status, significantly higher baseline frequency (%) of M-MDSCs was detected in 8 MSI-high EC patients (5.40% [3.94;8.70]) as compared to 18 MSS patients (1.19% [0.31; 2.64]) (p = 0.005) (supplementary Table 3). In Table 3, MSS patients had a significant increase Δ of 5.34% (95%CI − 8.11, − 2.56; p = 0.001) in M-MDSC at B2 as compared to MSI-high (Table 3). Tregs frequency significantly increased in CPA as compared to CP patients (0.67% [0.21; 0.87] vs 0.30% [0.18; 0.43], respectively), p = 0.038 (supplementary Table 4). Moreover, Treg subsets [naïve CD45RA+ FoxP3lo–(n-Tregs), activated/effector-CD45RA−FoxP3hi (a-Tregs), and not true Tregs CD45RA− FoxP3lo) (non-Tregs)] were evaluated. A significant increase in naïve-Tregs (n-Tregs) was detected in CPA compared to CP treated patients at B2 (0.07% [0.029; 0.140] vs 0.0150% [0.0007; 0.0300], respectively), p = 0.0044 (supplementary Table 5). Analysis of variations in circulating Treg subsets (Δ) showed modest but significant higher Δ a-Tregs (B1–B2 value) of 0.27% (95%CI − 0.55; − 0.001); p = 0.049) in MSS patients as compared to MSI-high, regardless of treatment type (Table 3).
Higher frequency of tumor infiltrated M-MDSC in MSS versus MSI-high ECs
To evaluate tumoral MDSC infiltration, 19/23 primary EC tissues, subgroup of peripheral analyzed patients molecularly characterized, were subjected to multiplex immunofluorescence for M-MDSC (CD11b+CD14+). Interestingly, although not significant, human CD11b+ and CD14+ stained cells were highly represented in MSS (n = 14) tumors as compared to MSI-high (n = 5). The median cell/mm2 [IQR] was 45.00[33.00, 69.00] in MSS vs 29 [15.00, 45.00] in MSI-high (Fig. 2).

EC patients displayed high tumor-infiltrating M-MDSCs in MSS compared to MSI-high. A Upper panel: Multiplex immunofluorescence (mIF) for M-MDSC (CD11b+CD14+) in primary tumor FFPE sections from EC-MITO-END3 patients. Box and whisker plot. Limits of boxes are 25th and 75th percentiles, with the thick line across the boxes being the median value. Whiskers indicate either the minimum and maximum values or a distance of 1.5 times the interquartile range from the edge of the box (whichever distance is smaller). Lower panel: microphotographs representing two cases of MSS and MSI-high primary tumors (original magnification 400× field); B peripheral pretreatment M-MDSC, (from supplementary Table 3) Box and whisker plot of peripheral
Lower CD4+ and higher M-MDSC cells were detected at baseline in peripheral blood of EC patients. Post hoc analysis.
CD4, Tregs, and MDSC were evaluated at baseline in 29 ECs versus 23 HDs. Lower frequency (%) of CD4+ cells (p < 0.005) and higher (%) of M-MDSCs (CD14+HLA-DR−/low-) (p = 0.020) were detected in EC patients as compared to HDs (Supplementary Fig. 4). Tregs or Tregs subset showed no significant variation in distribution between ECs and HDs (Supplementary Fig. 4).
Discussion
In the present manuscript, 29 EC patients participating to the MITO-END3 study were evaluated for the peripheral frequency of MDSCs and Tregs at pretreatment (B1) and after 6 cycle (B2) of assigned treatment, either CP (carboplatin/paclitaxel) or CPA (carboplatin/paclitaxel/avelumab).
At B2, peripheral effector Tregs frequency was significantly higher in patients treated with CPA while both treatments induced significant decrease in peripheral M-MDSC (− 5.41%) in TCGA 2-MSI-high as compared to TCGA-category 4 tumors (p = 0.004). In accordance, both treatments induced M-MDSCs (+ 5.34%) in MSS patients as compared to MSI-high patients (p = 0.001). Moreover, in 19/23 primary tumors higher frequency of M-MDSC was associated with MSS as compared to MSI-high ECs patients. Accordingly, previous data showed MDSC increase in advanced EC correlating with poor response to cytotoxic treatments [37].
In EC patients, MDSC were reported to correlate with peripheral blood MDSC and few CD8+ cells/abundant MDSCs represent independent prognostic factors [54]. MSS tumors are characterized by few TILS as compared to POLE and MSI tumors [55], although tumor/peritumor-infiltrating CD3+ and CD8+ lymphocytes did not significantly associate with objective response, mutational status, TMB, MSI in MITO-END3 study [23]. In the RUBY trial, though pMMR-MSS-ECs are considered poorly responder, dostarlimab benefit was also demonstrated in the pMMR/MSS patients, suggesting that the molecular subtype may be insufficient to stratify patients for immunotherapy [20]. Insights into TME can reveal “hot” tumors in dMMR or MSI-high and “cold” tumors inMMRp or MSS with limited benefit or resistance to ICIs [20].
High frequency of peripheral Tregs, in particular of naïve Treg subset, was detected in avelumab treated EC patients (CPA) at the end of cycle 6 of therapy. This evidence can be interpreted as a general biological effect of avelumab or as prognostic biomarker. Although previous data reported Tregs expansion in peripheral blood after ICI treatment [49], further investigation is needed to understand the interplay between avelumab, Tregs, and treatment outcomes. In preclinical models, tumoral Tregs increased after anti-PD-1(L1) [56,57,58] modifying the cyto/chemokines that regulate Tregs tumor recruitment [59,60,61] and associate with primary or secondary ICIs resistance [48, 50, 62]. Herein, regardless of treatment, increase in activated/effector-type Tregs associated with MSS suggesting multiple immune inhibitory networks in the MSS ECs-TME. Anti PD-(L)1 promoted tumor progression and concomitantly higher frequency of Tregs was reported [63,64,65]. Tiragolumab, an anti-TIGIT antibody Treg inhibitor, combined with atezolizumab demonstrated improved outcomes in NSCLC) [66, 67]. Interestingly, tazemetostat, an EZH2 inhibitor that impairs differentiation of n-Tregs to eTregs [68], is actually evaluated in ARID1A mutated ovarian/endometrial cancer.
Conclusion
Despite limited by patient number and lack of direct correlation between peripheral MDSCs/Tregs and clinical response, this is the first report showing association between longitudinal MDSC/Tregs and molecular characterization. With additional validation, these findings could enrich the selection of patients that may benefit from new treatment combinations.
Data availability
The core dataset from this study is available online on ZENODO (RRID:SCR_004129) (https://zenodo.org/records/15019459). Full data will be shared upon publication after a reasonable request to the corresponding author. Some deidentified individual participant data (i.e., baseline patient characteristics, treatment data, safety data, and follow-up data) will be available for sharing, with no time limit.
References
Crosbie EJ, Kitson SJ, McAlpine JN et al (2022) Endometrial cancer. Lancet 399:1412–1428. https://doi.org/10.1016/S0140-6736(22)00323-3
Lortet-Tieulent J, Ferlay J, Bray F et al (2017) International patterns and trends in endometrial cancer incidence, 1978–2013. JNCI J Natl Cancer Inst 110:354–361. https://doi.org/10.1093/jnci/djx214
Rahib L, Wehner MR, Matrisian LM et al (2021) Estimated projection of US cancer incidence and death to 2040. JAMA Netw Open 4:e214708. https://doi.org/10.1001/jamanetworkopen.2021.4708
Wagner VM, Backes FJ (2023) Do not forget about hormonal therapy for recurrent endometrial cancer: a review of options, updates, and new combinations. Cancers 15:1799
Miller DS, Filiaci VL, Mannel RS et al (2020) Carboplatin and paclitaxel for advanced endometrial cancer: final overall survival and adverse event analysis of a phase III trial (NRG Oncology/GOG0209). J Clin Oncol 38:3841–3850. https://doi.org/10.1200/jco.20.01076
Rütten H, Verhoef C, van Weelden WJ et al (2021) Recurrent endometrial cancer: local and systemic treatment options. Cancers 13:6275
Rousset-Rouviere S, Rochigneux P, Chrétien A-S et al (2021) Endometrial carcinoma: immune microenvironment and emerging treatments in immuno-oncology. Biomedicines 9:632
Levine DA, Getz G, Gabriel SB et al (2013) Integrated genomic characterization of endometrial carcinoma. Nature 497:67–73. https://doi.org/10.1038/nature12113
Zheng W (2023) Molecular classification of endometrial cancer and the 2023 FIGO staging: exploring the challenges and opportunities for pathologists. Cancers 15:4101
O’Hara AJ, Le Gallo M, Rudd ML et al (2020) High-resolution copy number analysis of clear cell endometrial carcinoma. Cancer Genet 240:5–14. https://doi.org/10.1016/j.cancergen.2019.10.005
Gargiulo P, Della Pepa C, Berardi S et al (2016) Tumor genotype and immune microenvironment in POLE-ultramutated and MSI-hypermutated Endometrial Cancers: new candidates for checkpoint blockade immunotherapy? Cancer Treat Rev 48:61–68. https://doi.org/10.1016/j.ctrv.2016.06.008
Concin N, Matias-Guiu X, Vergote I et al (2021) ESGO/ESTRO/ESP guidelines for the management of patients with endometrial carcinoma. Int J Gynecol Cancer 31:12–39. https://doi.org/10.1136/ijgc-2020-002230
Han KH, Park N, Lee M et al (2024) The new 2023 FIGO staging system for endometrial cancer: what is different from the previous 2009 FIGO staging system? J Gynecol Oncol. 35:e59
Similä-Maarala J, Soovares P, Pasanen A et al (2022) TCGA molecular classification in endometriosis-associated ovarian carcinomas: novel data on clear cell carcinoma. Gynecol Oncol 165:577–584. https://doi.org/10.1016/j.ygyno.2022.03.016
Stelloo E, Nout RA, Osse EM et al (2016) Improved risk assessment by integrating molecular and clinicopathological factors in early-stage endometrial cancer—combined analysis of the PORTEC cohorts. Clin Cancer Res 22:4215–4224. https://doi.org/10.1158/1078-0432.ccr-15-2878
Talhouk A, McConechy MK, Leung S et al (2015) A clinically applicable molecular-based classification for endometrial cancers. Br J Cancer 113:299–310. https://doi.org/10.1038/bjc.2015.190
Santoro A, Angelico G, Travaglino A et al (2021) New pathological and clinical insights in endometrial cancer in view of the updated ESGO/ESTRO/ESP guidelines. Cancers 13:2623
Eskander RN (2024) Revisiting immunotherapy in endometrial cancer. Clin Adv Hematol Oncol H&O 22:28–30
Westin SN, Moore K, Chon HS et al (2024) Durvalumab plus carboplatin/paclitaxel followed by maintenance durvalumab with or without olaparib as first-line treatment for advanced endometrial cancer: the phase III DUO-E trial. J Clin Oncol 42:283–299. https://doi.org/10.1200/jco.23.02132
Mirza MR, Chase DM, Slomovitz BM et al (2023) Dostarlimab for primary advanced or recurrent endometrial cancer. N Engl J Med 388:2145–2158. https://doi.org/10.1056/NEJMoa2216334
Lee SJ, Yoo JG, Kim JH et al (2025) Gynecologic oncology in 2024: breakthrough trials and evolving treatment strategies for cervical, uterine corpus, and ovarian cancers. J Gynecol Oncol. 36:e72
Colombo N, Biagioli E, Harano K et al (2024) Atezolizumab and chemotherapy for advanced or recurrent endometrial cancer (AtTEnd): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 25:1135–1146. https://doi.org/10.1016/s1470-2045(24)00334-6
Pignata S, Califano D, Lorusso D et al (2024) MITO END-3: efficacy of avelumab immunotherapy according to molecular profiling in first-line endometrial cancer therapy. Ann Oncol 35:667–676. https://doi.org/10.1016/j.annonc.2024.04.007
Wataru Y, Nobuyuki S, Hideo T et al (2011) Immunofluorescence-detected infiltration of CD4+FOXP3+ regulatory T cells is relevant to the prognosis of patients with endometrial cancer. Int J Gynecol Cancer 21:1628. https://doi.org/10.1097/IGC.0b013e31822c271f
Talhouk A, Derocher H, Schmidt P et al (2019) Molecular subtype not immune response drives outcomes in endometrial carcinoma. Clin Cancer Res 25:2537–2548. https://doi.org/10.1158/1078-0432.ccr-18-3241
Anderson Ana C, Joller N, Kuchroo Vijay K (2016) Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity 44:989–1004. https://doi.org/10.1016/j.immuni.2016.05.001
Sungu N, Yildirim M, Desdicioglu R et al (2019) Expression of immunomodulatory molecules PD-1, PD-L1, and PD-L2, and their relationship with clinicopathologic characteristics in endometrial cancer. Int J Gynecol Pathol 38:404–413. https://doi.org/10.1097/pgp.0000000000000543
Vanderstraeten A, Tuyaerts S, Amant F (2015) The immune system in the normal endometrium and implications for endometrial cancer development. J Reprod Immunol 109:7–16. https://doi.org/10.1016/j.jri.2014.12.006
Gabrilovich DI, Bronte V, Chen SH et al (2007) The terminology issue for myeloid-derived suppressor cells. Cancer Res 67:425. https://doi.org/10.1158/0008-5472.can-06-3037
Pan P-Y, Ma G, Weber KJ et al (2010) Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Can Res 70:99–108. https://doi.org/10.1158/0008-5472.CAN-09-1882
Pollard JW (2004) Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 4:71–78. https://doi.org/10.1038/nrc1256
Peña CG, Nakada Y, Saatcioglu HD et al (2015) LKB1 loss promotes endometrial cancer progression via CCL2-dependent macrophage recruitment. J Clin Investig 125:4063–4076. https://doi.org/10.1172/jci82152
Kumar V, Patel S, Tcyganov E et al (2016) The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol 37:208–220. https://doi.org/10.1016/j.it.2016.01.004
Weide B, Martens A, Zelba H et al (2014) Myeloid-derived suppressor cells predict survival of patients with advanced melanoma: comparison with regulatory T cells and NY-ESO-1- or melan-A–specific T cells. Clin Cancer Res 20:1601–1609. https://doi.org/10.1158/1078-0432.CCR-13-2508
Sasano T, Mabuchi S, Kozasa K et al (2018) The highly metastatic nature of uterine cervical/endometrial cancer displaying tumor-related leukocytosis: clinical and preclinical investigations. Clin Cancer Res 24:4018–4029. https://doi.org/10.1158/1078-0432.ccr-17-2472
Shimura K, Mabuchi S, Komura N et al (2021) Prognostic significance of bone marrow FDG uptake in patients with gynecological cancer. Sci Rep 11:2257. https://doi.org/10.1038/s41598-021-81298-1
Mabuchi S, Sasano T (2021) Myeloid-derived suppressor cells as therapeutic targets in uterine cervical and endometrial cancers. Cells 10:1073. https://doi.org/10.3390/cells10051073
Sakaguchi S, Sakaguchi N, Asano M et al (1995) Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 155:1151–1164
Miyara M, Yoshioka Y, Kitoh A et al (2009) Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 30:899–911. https://doi.org/10.1016/j.immuni.2009.03.019
Beriou G, Costantino CM, Ashley CW et al (2009) IL-17–producing human peripheral regulatory T cells retain suppressive function. Blood 113:4240–4249. https://doi.org/10.1182/blood-2008-10-183251
Kommoss S, McConechy MK, Kommoss F et al (2018) Final validation of the ProMisE molecular classifier for endometrial carcinoma in a large population-based case series. Ann Oncol 29:1180–1188. https://doi.org/10.1093/annonc/mdy058
Xi Z, Jing L, Le-Ni K et al (2019) Evaluation of PTEN and CD4+FOXP3+ T cell expressions as diagnostic and predictive factors in endometrial cancer: a case control study. Medicine 98:e16345. https://doi.org/10.1097/md.0000000000016345
Asaka S, Yen T-T, Wang T-L et al (2019) T cell-inflamed phenotype and increased Foxp3 expression in infiltrating T-cells of mismatch-repair deficient endometrial cancers. Mod Pathol 32:576–584. https://doi.org/10.1038/s41379-018-0172-x
Kolben T, Mannewitz M, Perleberg C et al (2022) Presence of regulatory T-cells in endometrial cancer predicts poorer overall survival and promotes progression of tumor cells. Cell Oncol 45:1171–1185. https://doi.org/10.1007/s13402-022-00708-2
Iurchenko NP, Glushchenko NM, Buchynska LG (2014) Comprehensive analysis of intratumoral lymphocytes and FOXP3 expression in tumor cells of endometrial cancer. Exp Oncol 36:262–266
Giatromanolaki A, Bates GJ, Koukourakis MI et al (2008) The presence of tumor-infiltrating FOXP3+ lymphocytes correlates with intratumoral angiogenesis in endometrial cancer. Gynecol Oncol 110:216–221. https://doi.org/10.1016/j.ygyno.2008.04.021
Guo F, Dong Y, Tan Q et al (2020) Tissue infiltrating immune cells as prognostic biomarkers in endometrial cancer: a meta-analysis. Dis Markers 2020:1805764. https://doi.org/10.1155/2020/1805764
van Gulijk M, van Krimpen A, Schetters S et al (2023) PD-L1 checkpoint blockade promotes regulatory T cell activity that underlies therapy resistance. Sci Immunol 8:eabn6173. https://doi.org/10.1126/sciimmunol.abn6173
Zappasodi R, Budhu S, Hellmann MD et al (2018) Non-conventional inhibitory CD4+Foxp3−PD-1hi T cells as a biomarker of immune checkpoint blockade activity. Cancer Cell 33:1017–32.e7. https://doi.org/10.1016/j.ccell.2018.05.009
Gaißler A, Bochem J, Spreuer J et al (2023) Early decrease of blood myeloid-derived suppressor cells during checkpoint inhibition is a favorable biomarker in metastatic melanoma. J Immunother Cancer 11:e006802. https://doi.org/10.1136/jitc-2023-006802
Peng X, Lee J, Adamow M et al (2023) A topic modeling approach reveals the dynamic T cell composition of peripheral blood during cancer immunotherapy. Cell Rep Methods 3:100546. https://doi.org/10.1016/j.crmeth.2023.100546
Pignata S, Scambia G, Schettino C et al (2023) Carboplatin and paclitaxel plus avelumab compared with carboplatin and paclitaxel in advanced or recurrent endometrial cancer (MITO END-3): a multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol 24:286–296. https://doi.org/10.1016/S1470-2045(23)00016-5
Perfetto SP, Ambrozak D, Nguyen R et al (2006) Quality assurance for polychromatic flow cytometry. Nat Protoc 1:1522–1530. https://doi.org/10.1038/nprot.2006.250
Mise Y, Hamanishi J, Daikoku T et al (2022) Immunosuppressive tumor microenvironment in uterine serous carcinoma via CCL7 signal with myeloid-derived suppressor cells. Carcinogenesis 43:647–658. https://doi.org/10.1093/carcin/bgac032
Bounous VE, Ferrero A, Campisi P et al (2022) Immunohistochemical markers and TILs evaluation for endometrial carcinoma. J Clin Med 11:5678
Kuczynski EA, Krueger J, Chow A et al (2018) Impact of chemical-induced mutational load increase on immune checkpoint therapy in poorly responsive murine tumors. Mol Cancer Ther 17:869–882. https://doi.org/10.1158/1535-7163.mct-17-1091
Ngiow SF, Young A, Jacquelot N et al (2015) A threshold level of intratumor CD8+ T-cell PD1 expression dictates therapeutic response to anti-PD1. Can Res 75:3800–3811. https://doi.org/10.1158/0008-5472.can-15-1082
Homet Moreno B, Zaretsky JM, Garcia-Diaz A et al (2016) Response to programmed cell death-1 blockade in a murine melanoma syngeneic model requires costimulation, CD4, and CD8 T Cells. Cancer Immunol Res 4:845–857. https://doi.org/10.1158/2326-6066.CIR-16-0060
Marshall LA, Marubayashi S, Jorapur A et al (2020) Tumors establish resistance to immunotherapy by regulating Treg recruitment via CCR4. J Immunother Cancer 8:e000764. https://doi.org/10.1136/jitc-2020-000764
Choi J, Lee HJ, Yoon S et al (2020) Blockade of CCL2 expression overcomes intrinsic PD-1/PD-L1 inhibitor-resistance in transglutaminase 2-induced PD-L1 positive triple negative breast cancer. Am J Cancer Res 10:2878–2894
Quagliariello V, Passariello M, Di Mauro A et al (2022) Immune checkpoint inhibitor therapy increases systemic SDF-1, cardiac DAMPs Fibronectin-EDA, S100/Calgranulin, galectine-3, and NLRP3-MyD88-chemokine pathways. Front Cardiovasc Med 9:930797. https://doi.org/10.3389/fcvm.2022.930797
Zhulai G, Oleinik E (2022) Targeting regulatory T cells in anti-PD-1/PD-L1 cancer immunotherapy. Scand J Immunol 95:e13129. https://doi.org/10.1111/sji.13129
Wakiyama H, Kato T, Furusawa A et al (2022) Treg-dominant tumor microenvironment is responsible for hyperprogressive disease after PD-1 blockade therapy. Cancer Immunol Res 10:1386–1397. https://doi.org/10.1158/2326-6066.CIR-22-0041
Kamada T, Togashi Y, Tay C et al (2019) PD-1+/regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci 116:9999–10008. https://doi.org/10.1073/pnas.1822001116
Poschel DB, Klement JD, Merting AD et al (2024) PD-L1 restrains PD-1(+)Nrp1(lo) Treg cells to suppress inflammation-driven colorectal tumorigenesis. Cell Rep 43:114819. https://doi.org/10.1016/j.celrep.2024.114819
Cho BC, Abreu DR, Hussein M et al (2022) Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol 23:781–792. https://doi.org/10.1016/S1470-2045(22)00226-1
Guan X, Hu R, Choi Y et al (2024) Anti-TIGIT antibody improves PD-L1 blockade through myeloid and T(reg) cells. Nature 627:646–655. https://doi.org/10.1038/s41586-024-07121-9
Søndergaard JN, Tulyeu J, Priest D et al (2025) Single cell suppression profiling of human regulatory T cells. Nat Commun 16:1325. https://doi.org/10.1038/s41467-024-55746-1
Acknowledgements
None.
Funding
(1) m2/6-c “approccio integrato per la caratterizzazione prognostico-predittiva del microambiente tumorale nel cancro renale” funded by ministero della salute (n.659 del 20/08/2018) (2) AIRC_ID_24746 “New generation of cxcr4 antagonist: peptide r54 from biomolecular mechanism to first in man clinical study”—ig 24746 funded by AIRC (n.1018 del 08/10/2021). Stefania Scala is the recipient of these grants. All the founders had no role in research design, conduct, analysis, and manuscript production.
Author information
Authors and Affiliations
Contributions
S.S. and S.P. conceived and planned the experiments, acquired of the financial support and the resources of study. S.S. coordinated the responsibility for research planning and execution. C.D. wrote the original draft and with G.R., M.N., E.C., and G.S. conducted investigation process and data evidence/collection. E.C, A.A, and F.P. applied the statistical methods of analysis and synthesized study data. C.D., G.R., M.N., E.C., G.S., and C.M. created figures and tables. G.R., M.N., A.S., F.P., and S.S. designed the methodology. M.N.; A.S., D.R., R.A., D.C., C.S., S.C.C., C.P., M.D.N, and A.P. managed activities to annotate and maintain research data. S.S, S.P., and F.P. provided software. D.C., L.A., F.P., and S.P. conducted the supervision for the research activity planning. M–N., D.C., F.P., and S.S. contributed in validation of research outputs. All authors critically reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
The research protocol was approved by Human Ethical Committee registration number 9/17–16/19 referred to ClinicalTrials.gov (NCT03503786) and EudraCT (2016–004403–31). The protocol for blood samples from healthy donors was approved by Institutional Review Boards CE 36/22 oss n.1177.
Consent to participate
Informed consent was obtained from all subjects involved in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
D’Alterio, C., Rea, G., Napolitano, M. et al. Association of peripheral monocytic myeloid-derived suppressor cells with molecular subtypes in single-center endometrial cancer patients receiving carboplatin + paclitaxel/avelumab (MITO-END3 trial). Cancer Immunol Immunother 74, 172 (2025). https://doi.org/10.1007/s00262-025-04021-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00262-025-04021-3