
Abstract
Chimeric antigen receptor (CAR) T cell therapy has emerged as a groundbreaking immunotherapy, demonstrating significant efficacy in treating B cell malignancies. In the context of multiple myeloma (MM), B cell maturation antigen (BCMA) has been identified as a critical target, driving the development of CAR T cell therapies designed to address this plasma cell cancer. Various CAR designs, utilizing different BCMA recognition domains, have yielded promising clinical results, leading to the approval of two BCMA-targeting CAR T cell therapies by the US Food and Drug Administration (FDA) for the treatment of MM. This review uniquely examines the BCMA CAR T cell landscape, emphasizing the design of recognition domains, clinical efficacy, and patient outcomes. It critically addresses emerging challenges such as antigen escape and toxicity profiles, which have surfaced alongside therapeutic advances. Moreover, the review spotlights cutting-edge developments, including dual-targeting CAR T strategies, advancements in CAR T cell manufacturing, and innovative allogeneic CAR T approaches utilizing healthy donor cells. By detailing both the breakthroughs and ongoing challenges in BCMA CAR T cell therapy, this review offers a comprehensive perspective on the current state and future possibilities of CAR T cell therapy for MM and its expanding role in treating hematologic malignancies and beyond.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Multiple myeloma (MM) is a clonal malignancy affecting plasma cells, primarily within the bone marrow (BM). Ranking as the second most common hematological cancer in the USA, it is anticipated that around 35,730 new cases will be diagnosed in 2023 [1]. Despite notable strides in treatment, MM remains an incurable disease, marked by recurring and increasingly refractory relapses [2]. Adoptive T cell therapy (ACT), a subset of immunotherapy, involves the extraction of T cells from a patient’s blood or tumor tissue. These cells are then expanded and modified ex vivo before being reintroduced into the patient [3]. ACT, employing engineered T cells expressing chimeric antigen receptors (CARs), has garnered FDA approval for treating patients with acute lymphoblastic leukemia (ALL), non-Hodgkin lymphoma (NHL), and MM [4, 5].
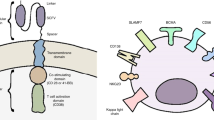
CARs typically comprise an extracellular antigen recognition domain linked to a hinge and transmembrane (TM) domain for cell surface anchoring, accompanied by a signaling endodomain [3] (Fig. 1). Traditionally, the extracellular antigen-binding domain in most CARs has been a single-chain variable fragment (scFv). This scFv is derived from the variable heavy chain (VH) and variable light chain (VL) regions of an antigen-specific monoclonal antibody (mAb), connected by a flexible linker (Fig. 1). The scFv enables CARs to recognize and bind to the target antigen with high specificity and affinity [3, 6]. Despite their advantages, scFvs may exhibit inefficient folding and a potential for aggregation. Various alternative options are available [7], such as camelid-derived single-domain antibody fragments (VHHs) [8], natural receptors or ligands [9], and scaffold molecules for extracellular antigen recognition domains [10] (Fig. 1). In this review, we provide an in-depth update on the clinical applications of various extracellular antigen recognition domain-based representative CARs in MM, with a particular focus on B cell maturation antigen (BCMA) as a critical target. We also explore the evolving landscape of CAR T cell therapy, highlighting the latest insights into emerging toxicities unique to CAR therapies, advances in bispecific CARs that aim to enhance targeting precision, and innovative approaches in CAR T cell manufacturing that address scalability and consistency challenges. Additionally, we examine the development of next-generation allogeneic CAR T therapies, underscoring their potential to broaden accessibility and improve therapeutic outcomes. By addressing these key areas, this review not only summarizes current advancements but also provides a forward-looking perspective on the future of CAR T cell therapies in MM.

Diverse extracellular recognition domains in BCMA CARs. A Classical BCMA CAR: Comprising an extracellular targeting domain derived from the variable regions of antibodies (scFv), a transmembrane domain (TM) for membrane anchoring, a flexible hinge domain, and intracellular signaling domains like CD3ζ and co-stimulatory domains such as CD28 or 4-1BB. These components collectively empower engineered T cells to identify and initiate an immune response against specific cancer cells. B Alternative BCMA CARs: Employ distinct extracellular antigen recognition domains, including VHH, Centyrin, natural ligands/receptors, and D-domains
BCMA as a CAR target
Foremost among considerations in advancing CAR T therapy is the identification of a tumor-specific antigen (TSA), which would be exclusively expressed on cancer cells and entirely absent from normal tissues [3, 11]. Such TSAs represent the ideal targets, as they could enable precise targeting without compromising healthy cells. However, TSAs are rare in MM and many other cancers, and this scarcity often necessitates the use of tumor-associated antigens (TAAs) as a compromise. While TAAs are not entirely exclusive to tumors, they typically exhibit higher expression on cancer cells than on normal tissues. This balance between therapeutic efficacy and minimizing “on-target, off-tumor” toxicity is essential in optimizing CAR T cell therapies for both safety and effectiveness [3, 12]. In the realm of MM, one extensively targeted TAA is BCMA (CD269), also known as tumor necrosis factor receptor superfamily member 17 (TNFRSF17) [13]. BCMA, a transmembrane glycoprotein, is notably present on differentiated plasma B cells in normal lymphoid tissues like the bone marrow, spleen, lymph nodes, and tonsils. Crucially, it lacks expression on naive B cells or other hematopoietic cells, including T cells, macrophages, neutrophils, and normal tissues [13,14,15]. BCMA interacts with two distinct agonist ligands: B cell-activating factor (BAFF) and a proliferation-inducing ligand (APRIL). These ligands are primarily secreted by bone marrow stromal cells, osteoclasts, and macrophages in a paracrine manner within the bone marrow [14,15,16]. Of note, APRIL exhibits higher affinity binding to BCMA compared to BAFF and also binds to transmembrane activator and calcium-modulator and cyclophilin ligand (TACI) (Fig. 2), while BAFF displays greater selectivity toward BAFF receptor (BAFFR) [14,15,16]. It is worth mentioning that TACI is also expressed on MM cells [17] (Fig. 2). The interaction between BCMA, APRIL, and TACI has garnered significant interest in CAR design (Fig. 1)., which will be discussed below [17]. Importantly, BCMA upregulation promotes MM cell growth, while its downregulation inhibits MM cell proliferation, rendering it an attractive target for CAR T cell therapy [18]. Additionally, the presence of soluble BCMA (sBCMA) in serum has been proposed as a potential biomarker in MM [19, 20], though it may pose a challenge to the full anti-BCMA CAR T cell activity.

Proposed designs of dual CARs targeting two multiple myeloma (MM)-associated antigens simultaneously. A MM cells express multiple known tumor-associated antigens providing opportunities for bispecific CAR design; BCMA and TACI are among these antigens, and they can be bound by soluble APRIL. B Bispecific CAR T Cells (Bi-CART) involve the individual transduction of T cells with two distinct CARs, achieved through separate vectors or a bicistronic vector. This process introduces two different CARs, with bivalent CARs manifesting in tandem or loop structures. In the tandem configuration, the variable light chain (VL) and variable heavy chain (VH) of one single-chain variable fragment (scFv) are directly linked to the VL-VH or VH-VL of another scFv. Conversely, the loop structure(Loop CAR T) is attained by rearranging the sequence of VL-VH or VH-VL from two scFvs
BCMA CAR T cell therapy
Significant efforts have been made to develop CARs using various classes of recognition domains targeting BCMA. This review will not cover all BCMA CARs but will focus on representative constructs from each class, as described below. Many of these CARs are currently under clinical evaluation, and two have been approved by the FDA. However, there is no consensus on the optimal design for BCMA CARs, and no in vivo studies or clinical trials have directly compared the performance of these different CAR T therapies. Consequently, there is no direct evidence to suggest that one class of BCMA CAR is superior to another.
Drawing from clinical experience with CD19 CAR therapy, some patients who relapsed after receiving murine CD19 CAR (mCD19 CAR) T cells did not respond to a second infusion of the same mCD19 CAR-T cells but achieved durable remissions with long-term persistence following treatment with humanized CD19 CAR-T cells [21,22,23,24]. This observation suggests that patients with MM may similarly benefit from sequential infusions of different classes of BCMA CAR-T cells to address therapeutic resistance or relapse. Currently, there are no clear guidelines on which BCMA CAR-T product to select for retreatment. The choice may depend on the underlying cause of the initial CAR T therapy failure. Implementing such a strategy has the potential to enhance treatment outcomes and significantly improve long-term patient survival.
BCMA scFv CAR
ScFvs represent the predominant ectodomains employed in CARs [25]. Among these, the most advanced scFv-based CAR-T cell product, targeting BCMA, is recognized as bb2121 (idecabtagene vicleucel or ide-cel). bb2121 is a lentiviral vector-based CAR incorporating a murine anti-human BCMA scFv (in VL-linker-VH orientation), a 4-1BB co-stimulatory motif, and a CD3-zeta T cell activation domain [26] (Table 1). Based on the outcomes of a confirmatory single-arm phase II trial (NCT03361748, KarMMa), the FDA has approved bb2121 (brand name: Abecma) (www.abecmahcp.com) for treating adult patients with relapsed or refractory multiple myeloma (RRMM) who have undergone at least four prior lines of therapy, including an immunomodulatory drug (IMiD), a proteasome inhibitor (PI), and an anti-CD38 monoclonal antibody (mAb) [27,28,29]. Among the 140 patients enrolled in this trial [27, 28], 128 received a single dose of Abecma. At a median follow-up of 13.3 months, 73% (94 out of 128) of patients responded to the treatment, with 33% achieving a complete response (CR) or better. Minimal residual disease (MRD)-negative status (< 10–5 nucleated cells) was confirmed in 26% of treated patients (33 out of 128) and in 79% of the 42 patients who achieved a CR or better. The median progression-free survival (PFS) was 8.8 months (95% CI, 5.6–11.6). Cytokine release syndrome (CRS) was experienced by 107 patients (84%), with 7 of them (5%) encountering grade 3 or higher events. Neurotoxic effects, also known as immune effector cell-associated neurotoxicity syndrome (ICANS), were observed in 23 patients (18%), with 4 patients (3%) experiencing grade 3 effects. Analysis of cellular kinetics revealed the presence of CAR + T cells in 29 out of 49 patients (59%) at the 6-month mark after infusion, and in 4 out of 11 patients (36%) at the 12-month mark [27].
The Phase 3 study, KarMMa-3 (NCT03651128) [30, 31], provided compelling evidence supporting the clinical advantages of early utilization of BCMA CAR T cell therapy in the treatment of MM. In this study (Table 2), a total of 254 patients received Abecma therapy, while 132 patients received a standard regimen. At a median follow-up of 18.6 months, the results demonstrated that Abecma therapy significantly improved PFS compared to the standard regimen (13.3 months vs. 4.4 months; 95% CI 0.38–0.65; P < 0.001). Additionally, the response rate was higher in the Abecma group (71%) compared to the standard-regimen group (42%) (P < 0.001). Notably, CR was achieved in 39% of patients receiving Abecma therapy, while only 5% of patients in the standard-regimen group achieved CR. Adverse events of grade 3 or 4 were experienced by 93% of patients in the Abecma group and 75% of patients in the standard-regimen group. Among the 225 patients who received Abecma therapy, CRS occurred in 88% of patients, with 5% experiencing grade 3 or higher events. Investigator-identified ICANS were observed in 15% of patients, with 3% experiencing grade 3 or higher events. These findings highlight the significant efficacy of Abecma in improving PFS and achieving higher response rates, particularly complete responses, compared to the standard treatment regimen.
BCMA VHH CAR
An alternative approach to antigen recognition in CAR design is the use of VHHs (Fig. 1), also known as nanobodies [32,33,34]. VHHs are characterized by their small, stable, single-domain structure, which provides them with high affinity and specificity, similar to scFvs [35]. Additionally, VHHs can be easily humanized for therapeutic purposes [35, 36].
One notable BCMA CAR design employing this alternative approach is LCAR-B38M (JNJ-68284528, also known as JNJ-4528) CAR-T, which has garnered significant attention [37,38,39]. LCAR-B38M incorporates 4-1BB co-stimulatory domain and targets two epitopes of BCMA using two tandem VHH sequences (Fig. 1; Table 1). The use of VHHs as antigen recognition domains in CAR-T cell therapy offers several advantages, including their compact size, ease of engineering, and potential for improved targeting and therapeutic efficacy [40]. The development of LCAR-B38M showcases the potential of utilizing VHHs as a promising strategy for CAR-T cell therapy, particularly in the context of targeting BCMA for the treatment of MM and other BCMA-expressing malignancies [37].
The FDA has officially granted approval for LCAR-B38M, marketed under the brand name CARVYKTI, for use as a treatment for adults with RRMM. This approval is specifically intended for patients who have undergone at least four prior lines of therapies, which must include a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody. The basis for this approval is the comprehensive analysis of data from the pivotal CARTITUDE-1 study, a phase 1b/2 clinical trial (NCT03548207) conducted in the USA [41, 42]. Findings from the CARTITUDE-1 study revealed that a single dose of CARVYKTI led to rapid and profound responses in heavily treated MM patients. Notably, these responses showed remarkable durability and were accompanied by a manageable safety profile [42]. The study revealed an impressive overall response rate (ORR) of 97%, with a notable 67% of patients achieving stringent CR. Additionally, the 12-month progression-free survival (PFS) and overall survival (OS) rates stood at 77% and 89%, respectively. Despite these promising outcomes, it is crucial to recognize the occurrence of treatment-related adverse events (AEs). The most prevalent AEs associated with the therapy included CRS affecting 95% of patients, as well as neutropenia (95%), anemia (68%), and thrombocytopenia (60%). Remarkably, only 4% of patients experienced grade 3–4 CRS. Furthermore, CAR T cell neurotoxicity was observed in 20 patients (21%), with 9% classified as grade 3 or 4. The safety profile of CARVYKTI remained consistent across subgroups, showing no emergence of new safety concerns.
The inaugural randomized Phase 3 trial, CARTITUDE-4 (NCT04181827), assesses the efficacy and safety of CARVYKTI in adult patients with RRMM who have undergone one to three prior lines of therapy. The study compares CARVYKTI with standard-of-care (SOC) treatments PVd or DPd (pomalidomide, bortezomib, and dexamethasone [PVd] or daratumumab, pomalidomide, and dexamethasone [DPd]) (Table 2). A total of 419 patients were enrolled, with 208 receiving CARVYKTI and 211 receiving standard care [43]. At a median follow-up of 15.9 months (range from 0.1 to 27.3), the CARVYKTI group demonstrated a significant improvement in median PFS compared to the SOC group (not reached vs. 11.8 months, hazard ratio 0.26, 95% CI 0.18–0.38, P < 0.001). The 12-month PFS rate was 75.9% (95% CI 69.4–81.1) in the CARVYKTI group and 48.6% (95% CI 41.5–55.3) in the SOC group. Moreover, more patients in the CARVYKTI group achieved an overall response (84.6% vs. 67.3%), CR or better (73.1% vs. 21.8%), and absence of MRD (60.6% vs. 15.6%) compared to the SOC group. The number of deaths from any cause was 39 in the CARVYKTI group and 46 in the standard-care group (hazard ratio 0.78, 95% CI 0.5 to 1.2). However, most patients in both groups experienced grade 3 or 4 AEs during treatment. Among the 176 patients who received CARVYKTI in the as-treated population, 76.1% had CRS (1.1% grade 3 or 4, no grade 5), 4.5% had ICANS (all grade 1 or 2), 1 patient had movement and neurocognitive symptoms (grade 1), 9.1% had cranial nerve palsy (8.0% grade 2, 1.1% grade 3), and 2.8% had CAR-T-related peripheral neuropathy (2.3% grade 1 or 2, 0.6% grade 3). The latest update shows that patients treated with cilta-cel (n = 208) experienced a 59% reduction in the risk of disease progression or death compared to those treated with SOC regimens (www.carvyktihcp.com). Based on data from the CARTITUDE-4 study, CARVYKTI has obtained FDA approval for earlier treatment of adult patients with RRMM. This approval is extended to patients who have undergone at least one prior line of therapy, including a proteasome inhibitor (PI) and an immunomodulatory agent (IMiD), and who are refractory to lenalidomide (Revlimid).
Similar to the concept of VHH CARs, Lam et al. [44] engineered a CAR named FHVH33-CD8BBZ, which includes a fully human heavy-chain only variable domain (FHVH) along with 4-1BB and CD3ζ domains (Fig. 1; Table 1). T cells expressing FHVH33-CD8BBZ demonstrated comparable cytokine release, degranulation, and eradication of mouse tumors, mirroring the outcomes observed with a CAR that differed only in substituting FHVH33 for a scFv. The presence of 4-1BB played a crucial role in mitigating activation-induced cell death (AICD) and enhancing the survival of T cells expressing FHVH33-containing CARs, in contrast to CD28 costimulation. These findings suggest that heavy-chain-only anti-BCMA CARs are well-suited for evaluation in clinical trials. In the subsequent Phase 1 clinical trial [45], 90% of 20 assessable FHVH-BCMA-T treatments resulted in objective responses (OR). Twelve treatments achieved very good partial response (VGPR), CR, or stringent CR. Currently, 50% of patients maintain responses (0–80 weeks), including 7 out of 8 at the highest two dose levels (88%, median duration 20 weeks, range 0 + to 35 + weeks). Only 1 patient experienced progressive MM, requiring immediate intervention. Among 8 patients with high-risk cytogenetics, 7 showed ORs, with ongoing responses observed in 2 patients with TP53 mutations and 1 patient with t(4;14) translocation. In a recent update [46], twenty-five patients with relapsed MM were treated, achieving a stringent complete response rate (sCR) of 52%. The median PFS was 78 weeks. Among the 24 evaluable patients, 6 (25%) experienced grade 3 CRS, with no cases exceeding this severity. Most anti-MM activity occurred within 2–4 weeks following FHVH-T infusion, as indicated by reductions in key MM markers, including serum free light chains, urine light chains, and bone marrow plasma cells. CAR+cell levels in the blood peaked during this period of MM clearance, typically between 7 and 15 days post-infusion. Interestingly, C–C chemokine receptor type 7 (CCR7) expression on CD4+FHVH-T cells was correlated with peak blood levels of FHVH-T. Single-cell RNA sequencing revealed a shift toward a more differentiated state in FHVH-T cells after infusion. Anti-CAR antibody responses were detected in 4 out of the 12 patients assessed. Overall, FHVH-T exhibited potent, rapid, and durable anti-MM activity.
BCMA Centyrin CAR
Centyrins represent a novel class of alternative scaffold proteins derived from a consensus fibronectin domain [47]. Notably, they exhibit a smaller size and are currently under development as the next generation of biological therapeutics, including CARs [48]. Similar to scFv molecules, Centyrins possess a remarkable ability to bind protein targets with exceptional affinity and specificity [47]. They harness the target specificity akin to that of antibodies and can be rapidly engineered for new targets of interest using in vitro display techniques. One of the advantages of Centyrins lies in their ease of manufacturing, as they can be readily produced across multiple expression systems. Furthermore, significant efforts have been made to engineer Centyrins with low potential for immunogenicity. Moreover, Centyrins demonstrate the convenience of room temperature storage and exhibit monodispersity even at high protein concentrations [47]. Overall, Centyrins showcase tremendous promise as a cutting-edge platform for the development of next-generation biological therapeutics, including CARs [48, 49].
A Centyrin-based BCMA CAR (Fig. 1; Table 1), known as “CARTyrin,” has been successfully developed and manufactured using a non-viral piggyBac (PB) transposon-based system. Specifically, one of the CAR constructs, called P-BCMA-101, represents a novel second-generation CAR-T product. P-BCMA-101 incorporates a fully human anti-BCMA Centyrin, TM, a 4-1BB co-stimulatory motif, and a CD3ζ activation domain [48] (Table 1). During preclinical studies, the manufacturing process demonstrated a notable production of T stem cell memory cells (TCM), which holds the potential for enhanced therapeutic longevity. To assess its safety and efficacy, P-BCMA-101 underwent a phase 1/2 clinical trial (NCT03288493; PRIME) involving heavily pretreated patients with RRMM [50,51,52]. In the clinical study update [52], P-BCMA-101 has shown efficacy and low toxicity in treating 90 patients across various dose levels, both as a single agent and in combination with rituximab (Rit) and lenalidomide (Len). Patients, heavily pretreated with a median of 6 prior regimens, demonstrated a favorable safety profile with no dose-limiting toxicities. The addition of Rit or Len did not increase toxicity, and the most common AEs were expected CAR-T-related effects. The overall response rates with Rit and Len combinations were 73% and 71%, respectively, allowing outpatient treatment in 25% of patients (Table 2).
Based on the P-BCMA-101 data, the development of the allogeneic CAR-T, P-BCMA-ALLO1, has been initiated [53]. In the Phase 1 study [54], seven patients received P-BCMA-ALLO1, and four successfully completed the dose-limiting toxicity (DLT) evaluation, making them eligible for response assessment. Adverse events were mostly grade 1 and 2, with a single patient experiencing serious grade 3 febrile neutropenia. Notably, DLTs, CRS, and ICANS were not observed. Early responses, including VGPR, PR, and stable disease (SD), were evident as early as week 2, resulting in an ORR of 75%. These preliminary findings indicate a favorable toxicity profile and promising efficacy for P-BCMA-ALLO1.
BCMA ligand-receptor CAR
Significant progress has been achieved in the development of CARs based on natural receptor-ligand interactions [9, 55]. Using natural receptor or ligand-based CARs offers potential advantages, including limited immunogenicity when the extracellular domain extensively employs a full-length native protein.
In the BCMA-CAR design, the high-affinity ligand APRIL, which interacts with BCMA and TACI, has been utilized [56, 57] (Figs. 1, 2). The CAR recognition domain was constructed using the sequence encoding residues 116–250 of the APRIL canonical sequence (Uniprot 075888), serving as the BCMA and TACI binding domain [57]. Dual targeting BCMA and TACI on myeloma cells has demonstrated promising outcomes, potentially addressing challenges such as antigen escape and downregulation, which can affect the durability of CAR T cell therapy. In a xenograft model of MM in mice, treatment with APRIL-based CAR T cells resulted in tumor clearance [57]. A phase 1 trial (NCT03287804) investigating the treatment of relapsed or refractory MM patients with APRIL-based CARTs has been ongoing since 2017 [58]. Notably, no instances of ICANS were reported. Among the treated patients, the dose cohorts of ≥ 225 × 10e6 demonstrated an ORR of 43%, including PR (28%) and VGPR (14%). Due to limited efficacy data observed to date [59], APRIL CAR trials (NCT03287804) have stopped recruiting. This decision reflects the ongoing challenge of achieving sufficient therapeutic benefit with ligand-based CAR T cell constructs in MM, prompting a reevaluation of these strategies in favor of more promising approaches.
An optimized APRIL CAR design called TriPRIL CAR, incorporating three binding domains to target both BCMA and TACI, has been proposed [56, 60]. This design utilizes the natural trimeric conformation of APRIL. In vitro studies demonstrated that TriPRIL CAR T cells exhibited enhanced specific cell lysis and robust degranulation when cultured with BCMA/TACI-positive cells [56]. In an in vivo study using a high tumor-burden xenograft mouse model, TriPRIL CAR T cells demonstrated the remarkable ability to completely eliminate tumors. In contrast, the original non-trimeric CAR T cells only managed to stabilize the disease progression [56]. Moreover, the TriPRIL CAR T cells exhibited their potential by effectively eradicating BCMA-knockout MM cells. This finding emphasizes the significance of dual-targeting TACI in the context of MM when compared to traditional BCMA CAR T cells and non-trimeric CAR T cells [56]. These promising preclinical results have paved the way for a groundbreaking first-in-human clinical trial (NCT05020444) that is currently underway. An advantage of using only human sequences in CAR-T cell therapies based on APRIL is the potential to avoid an immune response against murine components. However, a published cautionary narrative has emerged regarding APRIL-CAR T cells [61]. While APRIL-CAR T cells in a trimeric ligand binding conformation initially triggered rapid antitumor responses, these effects proved unsustainable. Rapid downmodulation of BCMA was observed across both APRIL-based and antibody-based binding moieties. This BCMA reduction primarily resulted from BCMA internalization, with additional contributions from trogocytosis by CAR T cells [61]. This study sheds light on the mechanisms of CAR T cell failure in MM targeting BCMA and provides insights that may inform the design of more durable and effective treatment strategies.
BCMA D domain CAR
The utilization of a de novo designed α-helical bundle known as α3D has proven successful as a scaffold for generating targeting domains (D domains) in CAR design [62, 63] (Fig. 1). These D domains are relatively smaller in size compared to scFv, approximately one-third the size, and lack disulfide bonds or N-linked glycosylation. These characteristics make them well-suited as modular targeting agents [62]. Due to their amenable nature, D domains can be manipulated and developed using various recombinant expression systems. The α3D scaffold itself demonstrates rapid and high thermal stability, enabling it to tolerate amino acid substitutions introduced in combinatorial libraries [62]. Moreover, the predominantly α-helical structure of the D domain provides an opportunity to explore potential paratope-epitope interfaces that may not be accessible to beta-sheet and loop folds typically found in scFv structures.
CAR T cells based on the D-domain (dd-BCMA CAR) targeting BCMA and other antigens have been developed and investigated for their anti-tumor activity [62, 64] (Table 1). Both in vitro and in vivo studies using mouse models have demonstrated the effective anti-tumor responses of dd-BCMA CAR T cells (64). Promising results have emerged from a Phase 1 study conducted by Arcellx [65, 66] further highlighting the potential of dd-BCMA CAR T cells in treating MM. In this study [66], all patients who received dd-BCMA CAR T cells exhibited a response, with 75% (9 out of 12) achieving CR or sCR. Over time, the responses deepened, and at the last data cut (median follow-up of 14 months), 89% (8 out of 9) of evaluable patients achieved MRD negativity. These findings underscore the safety of dd-BCMA CAR T cells and the durability of responses in patients with relapsed or refractory MM. Manageable toxicities, including CRS and ICANS, were observed and resolved with standard management at both dose levels.
Arcellx issued an update on the enduring and substantial responses seen in its Phase 1 expansion study of CART-ddBCMA in patients with RRMM. As of June 2, 2023, of the 38 patients who received treatment, all had a median of 4 prior therapies, with 100% being triple-refractory and 68% penta-refractory [67]. The median follow-up was 22 months, and CAR+cells comprised 70% of the infused T cells. The ORR was 100%, with 76% achieving CR/sCR and 86% being MRD-negative. Durable responses were seen, including in high-risk patients, with an 18-month PFS rate of 67%. CRS occurred in 95% of patients (mostly grade ≤ 2), and immune effector cell-associated neurotoxicity syndrome (ICANS) in 18%, with no lasting effects. No off-tumor toxicity or delayed neurotoxicity was observed. Based on these results, a dose of 115 ± 10 × 106 cells was recommended for the phase 2 study. CART-ddBCMA demonstrated promising efficacy and manageable safety in this heavily pretreated patient population.
Combinational antigen recognition: dual CARs
Although CAR T cell therapy has transformed the treatment landscape for MM, only a minority of patients achieve long-term disease remission. The underlying reasons for CAR-T resistance and relapse are complex and multifactorial. A key factor contributing to relapse after CAR T cell therapy is the emergence of tumors with reduced or absent expression of the target antigen, enabling them to evade immune elimination, a phenomenon known as “antigen escape [68,69,70,71,72]. Instances of relapse following CAR T cell therapy have demonstrated diminished or lost BCMA expression [71, 72]. Therefore, strategies to maintain or enhance BCMA expression may promote more robust and sustained therapeutic responses in BCMA CAR T cell therapy, either alone or in combination with CARs targeting other antigens. One promising approach is the use of γ-secretase inhibitors (GSIs), which prevent the enzymatic cleavage of BCMA from the cell surface, thereby increasing BCMA density on malignant plasma cells and potentially improving CAR T cell efficacy [73, 74]. Studies in myeloma cell lines and patient-derived cells have shown that GSI treatment dose-dependently raises BCMA levels on cancer cell surfaces while reducing the soluble BCMA fragment that may act as a decoy for BCMA-targeted CAR T cells [74]. This treatment also enhances CAR T cell recognition of cancer cells in vitro, suggesting that combining a GSI with BCMA CAR T cells may effectively counteract antigen escape [74]. Notably, this combination appears well-tolerated, with crenigacestat boosting target antigen density and enabling deep responses in heavily pretreated MM patients, including those with and without prior BCMA-targeted therapies [75]. However, the associated toxicity and the durability of these responses require further validation in clinical studies.
Interestingly, following anti-BCMA bispecific T cell engager (TCE) treatment, a higher incidence of BCMA mutations with various mechanisms leading to antigen loss, which was more frequent than previously recognized [76, 77]. In contrast, after BCMA CAR T Abecma therapy, a rare biallelic loss of TNFRSF17 was observed at relapse only in 6% of cases [76]. These findings highlight the role of immunoselection in driving BCMA(-) or mutant clones, contributing to relapse after targeted therapies. Mutations in BCMA confer varying sensitivities to different anti-BCMA treatments, underscoring the critical importance of considering the dynamic tumor antigen landscape in the development and selection of targeted immunotherapies for MM. Nevertheless, combination therapies targeting additional MM antigens, may offer a more effective treatment strategy [78]. Beyond BCMA, various other antigens have been identified and researched as potential candidates for immunotherapy, particularly in the context of CAR T cell therapy for MM [79, 80]. These antigens encompass a diverse array of targets (Fig. 2A), such as CD138, G-protein-coupled receptor class C group 5 member D (GPRC5D), TACI, signaling lymphocytic activation molecule family 7 (SLAMF7), natural killer group 2 member D (NKG2D) ligands, CD229, and integrin β7. Developing CAR designs that target dual or multiple antigens becomes crucial as a strategy to address antigen escape and bolster the effectiveness of immunotherapies against MM (Fig. 2B).
Targeting BCMA/CD38
The majority of the dual CAR T cells are presently undergoing evaluations in early-phase clinical trials or preclinical investigations. A notable advancement in this area was made by Li et al. [81] conducted a pioneering clinical trial in this domain, exploring a BCMA/CD38 tandem single-stalk CAR. In a cohort of 22 patients, all of whom had undergone at least two prior lines of treatment, the trial demonstrated promising results with an ORR of 91% and a CR rate of 54.5%. In another phase I clinical trial [82], BM38 CAR-T cells, designed to target both BCMA and CD38, showed significant efficacy. Among 23 patients, 87% achieved a clinical response with MRD negativity (≤ 10−4 nucleated cells), and 52% reached a SCR at a median follow-up of 9.0 months (range, 0.5–18.5 months). CRS occurred in 87% of patients, primarily grade 1–2 (65%), with no neurotoxicity observed. The median PFS was 17.2 months. Notably, two relapsed patients maintained BCMA and CD38 expression on MM cells, and BM38 CAR-T cells remained detectable in 77.8% of evaluable patients at 9 months and 62.2% at 12 months. These findings indicate that bispecific BCMA and CD38 CAR-T cells are feasible, safe, and highly effective in patients with RRMM.
Targeting BCMA/CD19
This dual CAR design targets both mature myeloma cells via BCMA and myeloma precursor cells via CD19, addressing challenges encountered in previous studies that focused solely on CD19 targeting, which proved ineffective due to the lack of CD19 expression. Yan et al. [83] reported findings from a phase 2 study involving 21 RRMM patients, administering a combination of anti-CD19 and anti-BCMA CAR T cells. The patient group, with a median of 6 prior therapies (range, 4–17) and 3 patients (14%) having undergone autologous stem cell transplantation before CAR T cell therapy, showed safe and efficacious results. Response rates, including ORR, very good PR (VGPR) or higher, CR or higher, and MRD negativity, were noted at 95%, 81%, 57%, and 81%, respectively. For patients achieving VGPR or better, the median progression-free survival (PFS) was 8 months (NCT04162353). Additionally, a sequential infusion approach using CD19-CAR T followed by BCMA-CAR T cells has been explored for RRMM treatment [84]. In this study, ten patients were treated, with seven receiving autologous CAR T cells and three receiving allogeneic CAR T cells. After a median follow-up of 20 months, The ORR was 90%, with four patients achieving SCR. Hematological toxicities were the most common severe adverse events, and grade 1–2 CRS occurred in 90% of patients. Notably, three patients treated with autologous CAR T cells maintained PFS beyond two years, whereas allogeneic CAR T recipients experienced disease progression within two months. These findings support the potential of sequential CAR T cell infusions as a feasible, safe, and effective treatment strategy for RRMM. Furthermore, limited studies have shown that a second infusion of CD19 CAR T cells is feasible and can lead to durable responses in a subset of patients who received an increased CART2 dose along with lymphodepletion before CART1 [85]. Collectively, these data may inform the design of future CAR T cell clinical trials for patients who have previously experienced failure with CAR T cell immunotherapy.
Furthermore, Gracell Biotechnologies has recently secured FDA Orphan Drug designation for its innovative dual CAR T therapy targeting both BCMA and CD19 in the context of MM. This therapy aims to enhance response depth and efficacy in RMM patients. Previously presented data at ASCO and EHA 2021 [86] covered 19 patients, and the current update includes 9 additional patients, bringing the total to 28 across three different dose levels. From October 2019 to November 2021, 28 heavily pretreated RRMM patients (ages 27–76) with a median of 5 prior treatment lines participated in a single-arm, open-label, multicenter trial [87]. Most patients (89.3%) were high risk, with some having extramedullary disease, plasma cell leukemia, or primary refractory disease. GC012F was administered as a single infusion at three dose levels following 2–3 days of lymphodepletion. As of January 26, 2022, the median follow-up was 6.3 months. The overall response rate (ORR) varied by dose level: 100% in DL1, 80% in DL2, and 93.8% in DL3. Notably, 100% of MRD-assessable patients (27/27) achieved MRD negativity. Cytokine Release Syndrome (CRS) was mostly low grade, with no severe cases of CRS or ICANS observed. The therapy showed no significant differences in pharmacokinetics across dose levels, with long-term persistence up to 793 days. In an updated analysis with a longer median follow-up [88], 29 heavily pretreated RRMM patients (median age 27–76) who had received a median of 5 prior therapies were treated with GC012F across three dose levels. The overall response rate was 93.1%, with a stringent complete response rate of 82.8%, and 100% of patients achieved MRD negativity. The median duration of response was 37.0 months, and median progression-free survival was 38.0 months. CRS occurred in 86.2% of patients, mostly grade 2 or lower, with no cases of ICANS reported. GC012F showed consistent persistence across dose levels, and sBCMA levels decreased significantly post-infusion. These results affirm the therapy’s potential, and further clinical studies are planned to confirm its efficacy in RRMM. In a related phase I study (NCT04935580) in transplant-eligible, high-risk, newly diagnosed multiple myeloma (NDMM) patients, 22 patients treated with the GC012F CAR-T cells achieved a 100% ORR and 95.5% sCR rate, with all patients reaching MRD negativity [89]. Only low-grade CRS was observed in 27% of patients, with no severe CRS, neurotoxicity, or treatment-related deaths reported. Robust CAR T cell expansion and durable responses were observed, suggesting GC012F’s potential for frontline treatment in MM. Further clinical trials are planned to confirm these results and explore the therapy’s efficacy in broader patient populations.
Targeting BCMA/GPRC5D
The development of an optimized dual-targeted CAR construct, simultaneously addressing BCMA and GPRC5D, holds promise in preventing BCMA-mediated relapse in MM [90]. To determine the most effective strategy, CF de Larrea et al. [90] conducted a comparative analysis of subtherapeutic doses of various forms of dual-targeted cellular therapy. Three strategies were employed: (i)Bi-CAR T: creating and combining mono-targeted CAR T cells in parallel, (ii) Bicistronic CAR T: utilizing bicistronic constructs that express separate CARs from one vector, and (iii) Loop CAR T: adopting a dual-scFv “single-stalk” CAR configuration (Fig. 2B). In cases targeting BCMA-negative disease, both bicistronic and pooled methods demonstrated superior efficacy, whereas, in dual-antigen–expressing disease, the bicistronic approach proved more effective than the pooled method. Mechanistically, the expression of two CARs on a single cell heightened the strength of interactions between CAR T cells and target cells.
A single-arm, phase 1 trial at Xuzhou Medical University in China evaluated the safety and activity of anti-BCMA/GPRC5D bispecific CAR T cells in 21 patients with RRMM [91]. Patients received varying doses of CAR T cells, and the maximum tolerated dose was identified as 2.0 × 10^6 cells per kg. The most common severe side effects were hematological, with 71% of patients experiencing mild cytokine release syndrome. The overall response rate was 86%, with 62% achieving a complete response or better. The trial concluded that these bispecific CAR T cells have a good safety profile and promising efficacy.
Toxicity of BCMA CAR T cell therapy
In a manner reminiscent of the impact of CD19 CAR T cell therapy on patients with acute lymphoblastic leukemia (ALL) and/or non-Hodgkin lymphoma (NHL), AE such as CRS and ICANS are frequently observed in MM patients undergoing BCMA CAR T cell therapy as well [48, 49] [92]. A meta-analysis conducted in 2020 [25], encompassing 23 distinct BCMA CAR T cell studies comprising 640 patients, revealed that 80.3% (with a range of 69.0–88.2) of patients encountered CRS. Severe CRS (grade ≥ 3) manifested in 14.1% of patients (with a range of 9.6–20.4). ICANS emerged at a pooled rate of 10.5% (with a range of 6.8–16.0), displaying variations across diverse studies. Nevertheless, the occurrence of severe CRS and/or ICANS is relatively less frequent in MM patients compared to those affected by ALL or NHL. This difference might be attributed to compromised T cell fitness in heavily pretreated patients grappling with RRMM. Moreover, it is essential to remain vigilant regarding other “on-target, off-tumor” side effects during clinical practice.
In a more recent development, Oekelen et al. [93] documented a case study involving an individual with MM who developed a progressive movement disorder featuring parkinsonism-like characteristics approximately three months post-receiving a BCMA-targeted CAR-T cell infusion known as CARVYKTI. This disorder was associated with the presence of CAR-T cells in both the bloodstream and cerebrospinal fluid, as well as lymphocytic infiltration within the basal ganglia. The researchers identified BCMA expression on neurons and astrocytes within the patient’s basal ganglia. Furthermore, public transcriptomic datasets corroborated BCMA RNA expression within the caudate of normal human brains, indicating the potential for an “on-target” effect related to anti-BCMA therapy. However, extensive BCMA expression profiling in the brains of adult individuals demonstrated low levels of BCMA RNA expression in the striatum of young donors, with such levels dwindling substantially beyond the age of 30. Importantly, it should be noted that BCMA protein is absent in normal adult human brains, thus rendering “on-target” toxicity within the brain implausible. Nevertheless, these findings underscore the importance of meticulous neurological monitoring for patients undergoing BCMA-targeted T cell therapies.
Some patients who underwent CAR T cell therapies, including BCMA CAR T cells, have experienced long-lasting remissions lasting a decade or more. However, there have been reported instances of secondary T cell malignancies, such as CAR-positive lymphomas and leukemias in some patients [94, 95]. In January 2024, the FDA mandated drug manufacturers to include a safety label warning on CAR T cell products. It is imperative for clinicians overseeing individuals treated with CAR T cells to promptly report any new cancer occurrences and establish comprehensive protocols for monitoring each patient throughout and after treatment. This approach will contribute to a more thorough understanding of secondary cancers in this patient population.
Shortening CAR T cells manufacturing time
The conventional approach to generating autologous CAR T cells involves extracting T cells from a patient’s blood through leukapheresis, modifying them to express a CAR specific to a TAA, and then expanding these modified T cells ex vivo over a period of days to weeks [96, 97]. Recent efforts have focused on shortening the duration of CAR T cell expansion [98, 99], resulting in a shorter overall manufacturing time from vein to vein (Supplementary Figure). This advancement has demonstrated enhanced antitumor activity compared to CAR T cells cultivated over a longer period, providing an opportunity for patients with aggressive diseases to receive treatment more promptly [100, 101].
A recent breakthrough involves the production of functional CAR T cells within a mere 24 h from T cells sourced from peripheral blood, bypassing the need for T cell activation and ex vivo expansion through viral or non-viral methods. The ongoing studies of GC012F, generated using the FasTCAR platform [102, 103], are presently undergoing Phase I trials. The company asserts that CAR T cells created on this platform exhibit characteristics of youthfulness, and improved proliferation and persistence capabilities. This accelerated manufacturing process has the potential to heighten cell production efficiency, thereby reducing costs and enhancing treatment accessibility.
Another promising innovation is seen in PHE885, an autologous fully human BCMA CAR-T cell product crafted using the T-Charge platform [104, 105]. This approach trims down the ex vivo culture time to approximately 24 h, culminating in the production of the final product in less than two days. The entire process relies on in vivo expansion following CAR-T cell infusion. In an early clinical study, PHE885 displayed encouraging outcomes among patients with relapsed or refractory multiple myeloma, as part of a Phase I, multicenter, dose-escalation trial [104]. Among the fifteen patients evaluated for both efficacy and safety, those receiving doses of 5 × 106 or 14.3 × 106 CAR-T cells exhibited an impressive best ORR of 100%, with responses intensifying over time. Sperling et al. [105] provided an overview of the updated phase 1 results for PHE885. Notably, PHE885 demonstrated an ORR of 98% in the 43 patients evaluable for efficacy, including a 100% ORR and 42% CRR at the active 10E6 dose. This was accompanied by a 10% incidence of grade 3 cytokine release syndrome (CRS) and a 6% occurrence of grade 3 ICANS.
The advancements in CAR T cell therapy manufacturing and technology represent a significant leap forward, offering substantial promise in providing more effective and efficient treatment options for individuals dealing with hematological malignancies and other aggressive diseases.
Allogenic BCMA CARs
Autologous CAR T therapies face challenges in logistics and extended manufacturing processes due to the necessity of processing each patient’s cells individually. Furthermore, in certain situations, manufacturing may not be feasible. Nevertheless, advancements in T cell engineering, gene editing, and cell manufacturing technologies have paved the way for extending T cell-based therapies to diverse cell types in the future, including NK cells, macrophages, hematopoietic stem cells, and induced pluripotent stem cells (iPSCs) [106,107,108]. Embracing an allogeneic approach that utilizes engineered cells from healthy donors holds the potential to broaden patient access to these therapies through the provision of a cost-effective and readily available “off-the-shelf” product [109].
The incorporation of cells from healthy donors has been extensively explored in the context of BCMA CAR T therapy for MM. Bioengineering strategies, including gene-editing techniques to knock out major histocompatibility complex (MHC) and T cell receptor (TCR) expression, have been employed to mitigate potential risks associated with graft-versus-host toxicity (GVHD) and host rejection. As we discussed above, the allogenic BCMA CAR T product, P-BCMA-ALLO1, is generated through the integration of two key platform technologies: the non-viral PB DNA Modification System and the precise Cas-CLOVER™ (CC) Site-Specific Gene Editing System. The CC System is employed to KO the endogenous T cell receptor (TCR) and beta-2 microglobulin, reducing Major Histocompatibility Complex (MHC) class I expression. This targeted KO aims to prevent GVHD and minimize host-versus-graft rejection. The CC System efficiently edits resting T cells, maintaining a high percentage of TSCM cells without generating unwanted off-target mutations, a critical factor in allo product development. Similarly, another anti-BCMA allogenic CAR T product ALLO-715 is evaluated in ongoing Phase 1 trials (UNIVERSAL, NCT04093596) [110].
Beyond donor-derived immune effector cells, immune cells derived from iPSCs present a promising platform for adoptive cellular therapy [111]. iPSC-derived lymphocytes offer the advantage of being “off-the-shelf” and can be meticulously selected and genetically edited to produce a consistent tumor-specific immune cell product [112, 113]. These iPSC-derived effector cells demonstrate potent antitumor activity comparable to conventional CAR T cells while retaining their intrinsic phenotype, thereby mitigating concerns about GVHD.
Several investigations are currently underway on immune cells derived from induced pluripotent stem cells (iPSCs) for the treatment of hematologic malignancies. A noteworthy example involves preclinical studies featuring FT576 cells [114], an innovative dual-target CAR iPSC-derived natural killer (NK) cell designed to target both BCMA and CD38. These studies demonstrated formidable cytotoxic activity against myeloma cell lines. The exploration of allogeneic approaches, utilizing cells sourced from healthy donors, holds the potential for creating readily available, “off-the-shelf” products. This approach aims to address logistical challenges and improve patient access to these critical and life-saving therapies.
Summary and future perspectives
In summary, alongside the remarkable success of CD19-targeted CAR T cell therapy in treating leukemia and lymphoma, the emergence of BCMA CAR T cell therapy stands as another triumph in the field. Various designs based on BCMA CAR T therapy have demonstrated significant efficacy in the treatment of MM, marking another milestone in the advancement of immunotherapy for hematological malignancies. While these BCMA CARs, including the two FDA-approved BCMA-directed CAR T products, CARVYKTI and Abecma, target the same antigen through similar mechanisms, there are notable differences among them. These differences include CAR T construct design, dosing regimens, and the characteristics of the patient populations in pivotal clinical trials. Such distinctions may contribute to the observed variations in efficacy and safety profiles. However, it remains challenging to definitively determine if one CAR T product is superior to the other.
Despite a subset of patients encountering severe CRS and ICANS, the effective implementation of management strategies underscores the adaptability of this therapeutic approach. The rapid evolution of CAR T cell therapy within MM immunotherapy establishes a robust foundation for ongoing research endeavors. However, the tumor microenvironment (TME) presents a significant challenge to the effectiveness of CAR T cell therapy by hindering CAR T cell trafficking, disrupting their metabolic function, and fostering an immunosuppressive environment that leads to T cell exhaustion. In MM, the bone marrow microenvironment is particularly complex, promoting tumor growth, immune evasion, and drug resistance [115]. This environment is enriched with immunosuppressive cells such as osteoclasts (OCs), myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), regulatory T and B cells (Tregs, Bregs), tumor-associated neutrophils (TANs), and bone marrow stromal cells (BMSCs) [116]. These cells not only support MM cell survival and proliferation but also diminish the cytotoxicity of effector T cells, facilitating immune escape. Increased OCs in MM contribute to bone disease and tumor growth by releasing factors like RANKL, APRIL, and IL-6, while also exhibiting immunosuppressive effects. TAMs, MDSCs, and neutrophils further drive immune evasion, angiogenesis, and resistance to therapy. Strategies to overcome the barriers posed by the TME on CAR T cell therapy efficacy are essential.
Future studies are poised to refine critical aspects such as CAR T cell manufacturing, multiple antigens targeting, allogeneic CAR immune cell therapy development, and meticulous neurological monitoring in the clinic with the overarching goal of enhancing the safety and efficacy of CAR T cell treatments for RRMM. Beyond the realm of MM, the horizon of BCMA CAR T cell therapy extends ambitiously, encompassing therapeutic interventions for solid tumors, infectious diseases, organ transplantation, and autoimmune disorders. This broadening scope reflects the versatile potential of BCMA CAR T cell therapy to revolutionize treatments across various medical domains, offering promising avenues for addressing diverse health challenges.
Data availability
No datasets were generated or analyzed during the current study.
References
Siegel RL, Miller KD, Wagle NS, Jemal A (2023) Cancer statistics, 2023. CA Cancer J Clin 73:17–48
Bhatt P, Kloock C, Comenzo R (2023) Relapsed/refractory multiple myeloma: a review of available therapies and clinical scenarios encountered in myeloma relapse. Curr Oncol 30:2322–2347
Xie Y, Hu Y, Zhou N, Yao C, Wu L, Liu L, Chen F (2020) CAR T-cell therapy for triple-negative breast cancer: Where we are. Cancer Lett 491:121–131
Mikkilineni L, Kochenderfer JN (2021) CAR T cell therapies for patients with multiple myeloma. Nat Rev Clin Oncol 18:71–84
Braendstrup P, Levine BL, Ruella M (2020) The long road to the first FDA-approved gene therapy: chimeric antigen receptor T cells targeting CD19. Cytotherapy 22:57–69
Sterner RC, Sterner RM (2021) CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J 11:1–11
Zajc CU, Salzer B, Taft JM, Reddy ST, Lehner M, Traxlmayr MW (2021) Driving CARs with alternative navigation tools–the potential of engineered binding scaffolds. FEBS J 288:2103–2118
Xie YJ, Dougan M, Jailkhani N et al (2019) Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc Natl Acad Sci 116:7624–7631
Branella GM, Spencer HT (2021) Natural receptor-and ligand-based chimeric antigen receptors: strategies using natural ligands and receptors for targeted cell killing. Cells 11:21
Frigault MJ, Bishop MR, Rosenblatt J et al (2022) Phase 1 Study of CART-ddBCMA for the treatment of subjects with relapsed and refractory multiple myeloma. Blood 140:7439
Yan T, Zhu L, Chen J (2023) Current advances and challenges in CAR T-Cell therapy for solid tumors: tumor-associated antigens and the tumor microenvironment. Exp Hematol Oncol 12:14
Gross G, Eshhar Z (2016) Therapeutic potential of T cell chimeric antigen receptors (CARs) in cancer treatment: counteracting off-tumor toxicities for safe CAR T cell therapy. Annu Rev Pharmacol Toxicol 56:59–83
Cho S-F, Anderson KC, Tai Y-T (2018) Targeting B cell maturation antigen (BCMA) in multiple myeloma: potential uses of BCMA-based immunotherapy. Front Immunol 9:1821
Yu B, Jiang T, Liu D (2020) BCMA-targeted immunotherapy for multiple myeloma. J Hematol Oncol 13:1–24
O’Connor BP, Raman VS, Erickson LD et al (2004) BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med 199:91–98
Paul B, Rodriguez C, Usmani SZ (2022) BCMA-targeted biologic therapies: the next standard of care in multiple myeloma therapy. Drugs 82:613
Novak AJ, Darce JR, Arendt BK, Harder B, Henderson K, Kindsvogel W, Gross JA, Greipp PR, Jelinek DF (2004) Expression of BCMA, TACI, and BAFF-R in multiple myeloma: a mechanism for growth and survival. Blood 103:689–694
Tai Y-T, Acharya C, An G et al (2016) APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood J Am Soc Hematol 127:3225–3236
Shen Y, Liu J, Wang B et al (2023) Serum soluble BCMA can be used to monitor relapse of multiple myeloma patients after chimeric antigen receptor T-cell immunotherapy. Curr Res Transl Med 71:103378
Shah N, Chari A, Scott E, Mezzi K, Usmani SZ (2020) B-cell maturation antigen (BCMA) in multiple myeloma: rationale for targeting and current therapeutic approaches. Leukemia 34:985–1005
Cao J, Wang G, Cheng H et al (2018) Potent anti-leukemia activities of humanized CD19-targeted Chimeric antigen receptor T (CAR-T) cells in patients with relapsed/refractory acute lymphoblastic leukemia. Am J Hematol 93:851–858
Zhao Y, Liu Z, Wang X et al (2019) Treatment with humanized selective CD19CAR-T cells shows efficacy in highly treated B-ALL patients who have relapsed after receiving murine-based CD19CAR-T therapies. Clin Cancer Res 25:5595–5607
Myers RM, Li Y, Barz Leahy A et al (2021) Humanized CD19-targeted chimeric antigen receptor (CAR) T cells in CAR-naive and CAR-exposed children and young adults with relapsed or refractory acute lymphoblastic leukemia. J Clin Oncol 39:3044–3055
An L, Lin Y, Deng B et al (2022) Humanized CD19 CAR-T cells in relapsed/refractory B-ALL patients who relapsed after or failed murine CD19 CAR-T therapy. BMC Cancer 22:393
Roex G, Timmers M, Wouters K et al (2020) Safety and clinical efficacy of BCMA CAR-T-cell therapy in multiple myeloma. J Hematol Oncol 13:1–14
Friedman KM, Garrett TE, Evans JW, Horton HM, Latimer HJ, Seidel SL, Horvath CJ, Morgan RA (2018) Effective targeting of multiple B-cell maturation antigen–expressing hematological malignances by anti-B-cell maturation antigen chimeric antigen receptor T cells. Hum Gene Ther 29:585–601
Anderson J, Larry D, Munshi NC, Shah N et al (2021) Idecabtagene vicleucel (ide-cel, bb2121), a BCMA-directed CAR T cell therapy, in relapsed and refractory multiple myeloma: updated KarMMa results
Munshi NC, Anderson J, Larry D, Shah N et al (2020) Idecabtagene vicleucel (ide-cel; bb2121), a BCMA-targeted CAR T-cell therapy, in patients with relapsed and refractory multiple myeloma (RRMM): initial KarMMa results. Am Soc Clin Oncol. https://doi.org/10.1200/JCO.2020.38.15_suppl.8503
Munshi NC, Anderson LD Jr, Shah N et al (2021) Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med 384:705–716
Rodriguez-Otero P, Ailawadhi S, Arnulf B et al (2023) Ide-cel or standard regimens in relapsed and refractory multiple myeloma. N Engl J Med 388:1002–1014
Delforge M, Baz RC, Cavo M et al (2020) KarMMa-3: a phase 3 study of idecabtagene vicleucel (ide-cel, bb2121), a BCMA-directed CAR T cell therapy vs standard regimens in relapsed and refractory multiple myeloma. Blood 136:24–25
Han L, Zhang J-S, Zhou J et al (2021) Single VHH-directed BCMA CAR-T cells cause remission of relapsed/refractory multiple myeloma. Leukemia 35:3002–3006
Fan XF, Zhuang Q, Yang L, Hao J, Zhao D, Zhao Y, Wang P, Geng D (2019) Preclinical assessment of LCAR-B38M, a novel BCMA-targeting chimeric antigen receptor (CAR)-T cell therapy in relapsed/refractory multiple myeloma. Clin Lymphoma Myeloma Leuk 19:e160
Bao C, Gao Q, Li L et al (2021) The application of nanobody in CAR-T therapy. Biomolecules 11:238
Asaadi Y, Jouneghani FF, Janani S, Rahbarizadeh F (2021) A comprehensive comparison between camelid nanobodies and single chain variable fragments. Biomarker Res 9:1–20
Vincke C, Loris R, Saerens D, Martinez-Rodriguez S, Muyldermans S, Conrath K (2009) General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J Biol Chem 284:3273–3284
Zhao W-H, Liu J, Wang B-Y et al (2018) A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol 11:1–8
Wang B-Y, Zhao W-H, Liu J et al (2019) Long-term follow-up of a phase 1, first-in-human open-label study of LCAR-B38M, a structurally differentiated chimeric antigen receptor T (CAR-T) cell therapy targeting B-cell maturation antigen (BCMA), in patients (pts) with relapsed/refractory multiple myeloma (RRMM). Blood 134:579
Zhao W-H, Wang B-Y, Chen L-J et al (2022) Four-year follow-up of LCAR-B38M in relapsed or refractory multiple myeloma: a phase 1, single-arm, open-label, multicenter study in China (LEGEND-2). J Hematol Oncol 15:1–12
Safarzadeh Kozani P, Naseri A, Mirarefin SMJ, Salem F, Nikbakht M, Evazi Bakhshi S, Safarzadeh Kozani P (2022) Nanobody-based CAR-T cells for cancer immunotherapy. Biomarker Res 10:24
Usmani SZ, Martin TG, Berdeja JG et al (2022) Phase 1b/2 study of ciltacabtagene autoleucel, a BCMA-directed CAR-T cell therapy, in patients with relapsed/refractory multiple myeloma (CARTITUDE-1): two years post-LPI. Am Soc Clin Oncol 40:8028
Berdeja JG, Madduri D, Usmani SZ et al (2021) Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. The Lancet 398:314–324
San-Miguel J, Dhakal B, Yong K et al (2023) Cilta-cel or standard care in lenalidomide-refractory multiple myeloma. New Engl J Med 389:335
Lam N, Trinklein ND, Buelow B, Patterson GH, Ojha N, Kochenderfer JN (2020) Anti-BCMA chimeric antigen receptors with fully human heavy-chain-only antigen recognition domains. Nat Commun 11:283
Mikkilineni L, Manasanch EE, Vanasse D et al (2020) Deep and durable remissions of relapsed multiple myeloma on a first-in-humans clinical trial of T cells expressing an anti-B-cell maturation antigen (BCMA) chimeric antigen receptor (CAR) with a fully-human heavy-chain-only antigen recognition domain. Blood 136:50–51
Mikkilineni L, Natrakul DA, Lam N et al (2024) Rapid anti-myeloma activity by T cells expressing an anti-BCMA CAR with a human heavy-chain-only antigen-binding domain. Mol Ther 32:503–526
Jacobs SA, Diem MD, Luo J, Teplyakov A, Obmolova G, Malia T, Gilliland GL, O’Neil KT (2012) Design of novel FN3 domains with high stability by a consensus sequence approach. Protein Eng Des Sel 25:107–117
Hermanson DL, Barnett BE, Rengarajan S et al (2016) A novel BCMA-specific, centyrin-based CAR-T product for the treatment of multiple myeloma. Blood 128:2127
Zhong X, D’Antona AM (2021) Recent advances in the molecular design and applications of multispecific biotherapeutics. Antibodies 10:13
Costello CL, Gregory TK, Ali SA et al (2019) Phase 2 study of the response and safety of P-Bcma-101 CAR-T cells in patients with relapsed/refractory (r/r) multiple myeloma (MM)(PRIME). Blood 134:3184
Costello CL, Cohen AD, Patel KK et al (2020) Phase 1/2 study of the safety and response of P-BCMA-101 CAR-T cells in patients with relapsed/refractory (r/r) multiple myeloma (MM)(PRIME) with novel therapeutic strategies. Blood 136:29–30
Costello C, Derman BA, Kocoglu MH et al (2021) Clinical trials of BCMA-targeted CAR-T cells utilizing a novel non-viral transposon system. Blood 138:3858
Cranert SA, Richter M, Tong M, Weiss L, Tan Y, Ostertag EM, Coronella J, Shedlock DJ (2019) Manufacture of an allogeneic CAR-T stem cell memory product candidate for multiple myeloma, P-Bcma-ALLO1, is robust, reproducible and highly scalable. Blood 134:4445
Kocoglu M, Asch A, Ramakrishnan A et al (2022) 47P Phase I study to assess the safety and efficacy of P-BCMA-ALLO1: a fully allogeneic CAR-T therapy, in patients with relapsed/refractory multiple myeloma (RRMM). Immuno-Oncol Technol 16:100152
Murad JM, Graber DJ, Sentman CL (2018) Advances in the use of natural receptor-or ligand-based chimeric antigen receptors (CARs) in haematologic malignancies. Best Pract Res Clin Haematol 31:176–183
Schmidts A, Ormhøj M, Choi BD et al (2019) Rational design of a trimeric APRIL-based CAR-binding domain enables efficient targeting of multiple myeloma. Blood Adv 3:3248–3260
Lee L, Draper B, Chaplin N et al (2018) An APRIL-based chimeric antigen receptor for dual targeting of BCMA and TACI in multiple myeloma. Blood J Am Soc Hematol 131:746–758
Popat R, Zweegman S, Cavet J et al (2019) Phase 1 first-in-human study of AUTO2, the first chimeric antigen receptor (CAR) T cell targeting APRIL for patients with relapsed/refractory multiple myeloma (RRMM). Blood 134:3112
Lee L, Lim WC, Galas-Filipowicz D et al (2023) Limited efficacy of APRIL CAR in patients with multiple myeloma indicate challenges in the use of natural ligands for CAR T-cell therapy. J Immunother Cancer 11:e006699
Schmidts A, Ormhoj M, Taylor AO, Lorrey SJ, Scarfò I, Frigault MJ, Choi BD, Maus MV (2018) Engineering an optimized trimeric APRIL-based CAR to broaden targetability of multiple myeloma. Blood 132:2059
Camviel N, Wolf B, Croce G, Gfeller D, Zoete V, Arber C (2022) Both APRIL and antibody-fragment-based CAR T cells for myeloma induce BCMA downmodulation by trogocytosis and internalization. J Immunother Cancer 10:e005091
Qin H, Edwards JP, Zaritskaya L, Gupta A, Mu CJ, Fry TJ, Hilbert DM, LaFleur DW (2019) Chimeric antigen receptors incorporating D domains targeting CD123 direct potent mono-and bi-specific antitumor activity of T cells. Mol Ther 27:1262–1274
Buonato JM, Edwards JP, Zaritskaya L, Witter AR, Gupta A, LaFleur DW, Tice DA, Richman LK, Hilbert DM (2022) Preclinical efficacy of bcma-directed car T cells incorporating a novel d domain antigen recognition domain. Mol Cancer Ther 21:1171–1183
Buonato JM, Edwards J, Zaritskaya L, Gupta A, Richman L, Hilbert D, LaFleur D, Tice D (2021) Design and demonstration of potent in vitro and in vivo activity for CART ddBCMA, a BCMA targeted CAR T cell therapy incorporating a non scFv binding domain. In: ASGCT virtual meeting. Virtual
Frigault MJ, O’Donnell E, Raje NS et al (2021) Phase 1 Study of CART-ddBCMA, a CAR-T therapy utilizing a novel synthetic binding domain, for the treatment of subjects with relapsed and refractory multiple myeloma. Clin Lymphoma Myeloma Leukemia. https://doi.org/10.1016/S2152-2650(22)00276-2
Frigault MJ, Bishop MR, Rosenblatt J et al (2023) Phase 1 study of CART-ddBCMA for the treatment of subjects with relapsed and refractory multiple myeloma. Blood Adv 7:768–777
Frigault MJ, Rosenblatt J, Dhakal B et al (2023) Phase 1 study of CART-Ddbcma for the treatment of patients with relapsed and/or refractory multiple myeloma: results from at least 1-year follow-up in all patients. Blood 142:1023
Majzner RG, Mackall CL (2018) Tumor antigen escape from CAR T-cell therapy. Cancer Discov 8:1219–1226
Walsh Z, Ross S, Fry TJ (2019) Multi-specific CAR targeting to prevent antigen escape. Curr Hematol Malig Rep 14:451–459
Simon S, Riddell SR (2020) Dual targeting with CAR T cells to limit antigen escape in multiple myeloma. Blood Cancer Discov 1:130–133
Samur MK, Fulciniti M, Aktas Samur A et al (2021) Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat Commun 12:868
Lee H, Ahn S, Maity R et al (2023) Mechanisms of antigen escape from BCMA-or GPRC5D-targeted immunotherapies in multiple myeloma. Nat Med 29:2295
Green DJ, Pont M, Cowan AJ et al (2019) Response to Bcma CAR-T cells correlates with pretreatment target antigen density and is improved by small molecule inhibition of gamma secretase. Blood 134:1856
Pont MJ, Hill T, Cole GO et al (2019) γ-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood J Am Soc Hematol 134:1585–1597
Cowan AJ, Pont MJ, Sather BD et al (2023) γ-Secretase inhibitor in combination with BCMA chimeric antigen receptor T-cell immunotherapy for individuals with relapsed or refractory multiple myeloma: a phase 1, first-in-human trial. Lancet Oncol 24:811–822
Samur MK, Martin N, Thompson E, Fulciniti M, Aktas-Samur A, Kaiser S, Munshi NC (2022) Differences in single cells between BCMA-targeting CAR T-cell therapy responders and non-responders reveals initial resistance and acquired resistance are driven by different factors. Blood 140:2106–2107
Lee H, Ahn S, Maity R et al (2023) Mechanisms of antigen escape from BCMA-or GPRC5D-targeted immunotherapies in multiple myeloma. Nat Med 29:2295–2306
Li W, Zhang B, Cao W et al (2023) Identification of potential resistance mechanisms and therapeutic targets for the relapse of BCMA CAR-T therapy in relapsed/refractory multiple myeloma through single-cell sequencing. Exp Hematol Oncol 12:44
Ohmine K, Uchibori R (2022) Novel immunotherapies in multiple myeloma. Int J Hematol 115:799
Padda J, Khalid K, Zubair U, Peethala MM, Kakani V, Goriparthi L, Almanie AH, Cooper AC, Jean-Charles G (2021) Chimeric antigen receptor T cell therapy and its significance in multiple myeloma. Cureus. https://doi.org/10.7759/cureus.15917
Li C, Mei H, Hu Y et al (2019) A bispecific CAR-T cell therapy targeting Bcma and CD38 for relapsed/refractory multiple myeloma: updated results from a phase 1 dose-climbing trial. Blood 134:930
Mei H, Li C, Jiang H et al (2021) A bispecific CAR-T cell therapy targeting BCMA and CD38 in relapsed or refractory multiple myeloma. J Hematol Oncol 14:1–17
Yan Z, Cao J, Cheng H et al (2019) A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: a single-arm, phase 2 trial. Lancet Haematol 6:e521–e529
Yan L, Qu S, Shang J et al (2021) Sequential CD19 and BCMA-specific CAR T-cell treatment elicits sustained remission of relapsed and/or refractory myeloma. Cancer Med 10:563–574
Gauthier J, Bezerra ED, Hirayama AV et al (2021) Factors associated with outcomes after a second CD19-targeted CAR T-cell infusion for refractory B-cell malignancies. Blood J Am Soc Hematol 137:323–335
Jiang H, Dong B, Gao L et al (2021) Long-term follow-up results of a multicenter first-in-human study of the dual BCMA/CD19 Targeted FasT CAR-T GC012F for patients with relapsed/refractory multiple myeloma. J Clin Oncol 39:8014
Du J, Jiang H, Dong B et al (2022) Updated results of a multicenter first-in-human study of BCMA/CD19 dual-targeting fast CAR-T GC012F for patients with relapsed/refractory multiple myeloma (RRMM). J Cli Oncol 16:8005
Du J, Fu W, Jiang H et al (2023) Updated results of a Phase I, open-label study of BCMA/CD19 Dual-targeting FasTCAR-T GC012F for patients with relapsed/refractory multiple myeloma (RRMM). J Clin Oncol 16:8005
Du J, Qiang W, Lu J et al (2023) Updated results of a phase I open-label single-arm study of dual targeting BCMA and CD19 Fastcar-T cells (GC012F) as first-line therapy for transplant-eligible newly diagnosed high-risk multiple myeloma. Blood 142:1022
Fernández de Larrea C, Staehr M, Lopez AV et al (2020) Defining an optimal dual-targeted CAR T-cell therapy approach simultaneously targeting BCMA and GPRC5D to prevent BCMA escape–driven relapse in multiple myeloma. Blood Cancer Discovry 1:146–154
Zhou D, Sun Q, Xia J et al (2024) Anti-BCMA/GPRC5D bispecific CAR T cells in patients with relapsed or refractory multiple myeloma: a single-arm, single-centre, phase 1 trial. Lancet Haematol 11:751
Cohen AD, Parekh S, Santomasso BD et al (2022) Incidence and management of CAR-T neurotoxicity in patients with multiple myeloma treated with ciltacabtagene autoleucel in CARTITUDE studies. Blood Cancer J 12:32
Van Oekelen O, Aleman A, Upadhyaya B et al (2021) Neurocognitive and hypokinetic movement disorder with features of parkinsonism after BCMA-targeting CAR-T cell therapy. Nat Med 27:2099–2103
Elsallab M, Ellithi M, Lunning MA, D’Angelo C, Ma J, Perales M-A, Frigault M, Maus MV (2024) Second primary malignancies after commercial CAR T cell therapy: analysis of FDA adverse events reporting system (FAERS). Blood. 143:3099
Ghilardi G, Fraietta JA, Gerson JN et al (2024) T cell lymphoma and secondary primary malignancy risk after commercial CAR T cell therapy. Nat Med 30:984
Levine BL, Miskin J, Wonnacott K, Keir C (2017) Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev 4:92–101
Wang X, Rivière I (2016) Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncol 3:16015
Ghassemi S, Nunez-Cruz S, O’Connor RS et al (2018) Reducing ex vivo culture improves the antileukemic activity of chimeric antigen receptor (CAR) T cells. Cancer Immunol Res 6:1100–1109
Yang J, He J, Zhang X et al (2022) Next-day manufacture of a novel anti-CD19 CAR-T therapy for B-cell acute lymphoblastic leukemia: first-in-human clinical study. Blood Cancer J 12:104
Tu S, Huang R, Guo Z et al (2019) Shortening the ex vivo culture of CD19-specific CAR T-cells retains potent efficacy against acute lymphoblastic leukemia without CAR T-cell-related encephalopathy syndrome or severe cytokine release syndrome. Am J Hematol 94:E322–E325
Kebriaei P, Huls H, Neel SL et al (2017) Shortening the time to manufacture CAR+ T cells with sleeping beauty system supports T-cell engraftment and anti-tumor effects in patients with refractory CD19+ tumors. Blood 130:2060
Jiang H, Dong B, Gao L et al (2020) Clinical results of a multicenter study of the first-in-human dual BCMA and CD19 targeted novel platform fast CAR-T cell therapy for patients with relapsed/refractory multiple myeloma. Blood 136:25–26
Du J, Fu W, Lu J et al (2022) Phase I open-label single-arm study of BCMA/CD19 dual-targeting FasTCAR-T Cells (GC012F) as first-line therapy for transplant-eligible newly diagnosed high-risk multiple myeloma. Blood 140:889–890
Sperling AS, Nikiforow S, Nadeem O et al (2021) Phase I study of PHE885, a fully human BCMA-directed CAR-T cell therapy for relapsed/refractory multiple myeloma manufactured in< 2 days using the T-charge TM platform. Blood 138:3864
Sperling AS, Derman BA, Nikiforow S et al (2023) Updated phase I study results of PHE885, a T-Charge manufactured BCMA-directed CAR-T cell therapy, for patients (pts) with r/r multiple myeloma (RRMM). Am Soc Clin Oncol 41:8004
Townsend MH, Bennion K, Robison RA, O’Neill KL (2020) Paving the way towards universal treatment with allogenic T cells. Immunol Res 68:63–70
Aftab BT, Sasu B, Krishnamurthy J, Gschweng E, Alcazer V, Depil S (2020) Toward “off-the-shelf” allogeneic CAR T cells. Advances in Cell and Gene Therapy 3:e86
Kim DW, Cho J-Y (2020) Recent advances in allogeneic CAR-T cells. Biomolecules 10:263
Manier S, Ingegnere T, Escure G, Prodhomme C, Nudel M, Mitra S, Facon T (2022) Current state and next-generation CAR-T cells in multiple myeloma. Blood Rev 54:100929
Mailankody S, Liedtke M, Sidana S et al (2021) Universal updated phase 1 data validates the feasibility of allogeneic anti-BCMA ALLO-715 therapy for relapsed/refractory multiple myeloma. Blood 138:651
Nianias A, Themeli M (2019) Induced pluripotent stem cell (iPSC)–derived lymphocytes for adoptive cell immunotherapy: recent advances and challenges. Curr Hematol Malig Rep 14:261–268
Sadeqi Nezhad M, Abdollahpour-Alitappeh M, Rezaei B, Yazdanifar M, Seifalian AM (2021) Induced pluripotent stem cells (iPSCs) provide a potentially unlimited T cell source for CAR-T cell development and off-the-shelf products. Pharm Res 38:931–945
Kawamoto H, Masuda K, Nagano S (2021) Regeneration of antigen-specific T cells by using induced pluripotent stem cell (iPSC) technology. Int Immunol 33:827–833
Goodridge JP, Bjordahl R, Mahmood S et al (2020) FT576: multi-specific off-the-shelf CAR-NK cell therapy engineered for enhanced persistence, avoidance of self-fratricide and optimized mab combination therapy to prevent antigenic escape and elicit a deep and durable response in multiple myeloma. Blood 136:4–5
Zhang X, Zhang H, Lan H, Wu J, Xiao Y (2023) CAR-T cell therapy in multiple myeloma: current limitations and potential strategies. Front Immunol 14:1101495
Desantis V, Savino FD, Scaringella A, Potenza MA, Nacci C, Frassanito MA, Vacca A, Montagnani M (2022) The leading role of the immune microenvironment in multiple myeloma: a new target with a great prognostic and clinical value. J Clin Med 11:2513
Acknowledgements
The illustration materials utilized in this work were partially obtained from the Icon Library on BioRender.com.
Funding
This work was partially sponsored by the Natural Science Foundation of Shenzhen Science and Technology Innovation Commission (JCY20220530155613030).
Author information
Authors and Affiliations
Contributions
FC conceptualized the review and critically reviewed and edited the manuscript. YH, YX, XW, LY, HG, ZY, and LM wrote the first draft of the manuscript. All authors contributed critically to writing and editing the drafts and gave final approval for publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors have approved to publish this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hu, Y., Xie, Y., Wang, X. et al. Targeting BCMA in multiple myeloma: designs, challenges, and future directions. Cancer Immunol Immunother 74, 77 (2025). https://doi.org/10.1007/s00262-024-03913-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00262-024-03913-0