
Abstract
Fungal infections represent a serious global health threat. The new emerging pathogens and the spread of different forms of resistance are now hardly challenging the tools available in therapy and diagnostics. With the commonly used diagnoses, fungal identification is often slow and inaccurate, and, on the other hand, some drugs currently used as treatments are significantly affected by the decrease in susceptibility. Herein, the antifungal arsenal is critically summarized. Besides describing the old approaches and their mechanisms, advantages, and limitations, the focus is dedicated to innovative strategies which are designed, identified, and developed to take advantage of the discrepancies between fungal and host cells. Relevant pathways and their role in survival and virulence are discussed as their suitability as sources of antifungal targets. In a similar way, molecules with antifungal activity are reported as potential agents/precursors of the next generation of antimycotics. Particular attention was devoted to biotechnological entities, to their novelty and reliability, to drug repurposing and restoration, and to combinatorial applications yielding significant improvements in efficacy.
Key points
• New antifungal agents and targets are needed to limit fungal morbidity and mortality.
• Therapeutics and diagnostics suffer of delays in innovation and lack of targets.
• Biologics, drug repurposing and combinations are the future of antifungal treatments.
Similar content being viewed by others


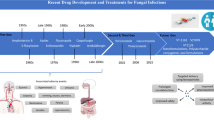
A newly found old threat
Even if part of the microbiological world, fungi have never had the consideration that bacteria, and viruses on the contrary received. The underestimation of their risk led to a minor interest from the scientific community hence, to less information and drugs currently available. Fungi share with their hosts many similarities proper of eukaryotic organisms, making the hunt for effective and safe antifungal drugs incredibly challenging from both the scientific and economic point of view. Moreover, several bad behaviors and practices have worsened the situation like the extensive and intensive use of antifungal agents in agriculture, or the incorrect use by patients. In response to our negligence, now we must face not just the great plague of fungal infections itself, but also the rise, reappearance, and spread of species carrying intrinsic and multidrug resistance. This review considers a well-known threat that is quickly becoming more dangerous. Our aim is not just giving a concise description of the state-of-the-art but offering a real insight toward the next generation of antifungal entities. We examined old and new treatments of natural, semi-/synthetic origin allowing great consideration to biotechnological agents. With a fresh point of view, we tried to find logical interrelationships among pathways and, therefore, among drug activity that affect different and interconnected players. We showed the options available and under investigation with extremely updated information and we condensed (when relevant and known) details that in previous works were only superficially or partially discussed like characteristics, mechanisms of action, spectra, pharmacokinetics, toxicities, positive and negative interactions, and, about that, combinatorial approaches and repurposing strategies. Finally, we also faced a hot topic represented by diagnoses that are affected by poor novelty, slowness, and inaccuracy—aspects that fall back into delayed therapies and negative outcomes in terms of patient survival.
Diagnosis
Diagnosis is an important issue, especially for fungal infections. Despite the development of new diagnostic agents and techniques, fungal identification relies on (from the most to the least used) microscope examination, microbial cultures, antigen detections, serological tests, and molecular analyses (Kozel and Wickes 2014). The first two processes are based on the growth of fungi in plates or liquid medium and are followed by susceptibility testing to find MICs and breakpoints for the correct use of antifungal drugs. Despite these procedures being still largely employed, they delay the beginning of the treatment and negatively affect patient recovery or survival. Moreover, the accuracy is lower compared to more recent techniques as molecular and immunological assays. However, about them, less information is available on online platforms, and databases are poor and limited to a few and more common mycoses (Zhang et al. 2021b). The presence in the market of diagnostic kits for antigen detection is also restricted, and sensitivity and specificity are not always optimal (Wickes and Wiederhold 2018; Rodrigues and Nosanchuk 2020). Finally, there is patient variability: symptoms are diversified from subject to subject even if the infection is caused by the same pathogen. In this regard, the degree of infection, comorbidity, and physio-pathological and pharmacological conditions like immunodeficiency or immunosuppressive treatments are associated with the fastest progressions and worst outcomes. This sad view highlights the urgent need for not just new antifungal drugs but also for new data and techniques for clear, easy, quick-to-perform, robust, sensitive, and specific diagnoses and diagnostic tools.
Therapy
The old
The battle against bacteria continually met fast times of new target identifications and compound developments. Conversely, the hunt for antifungal drugs was characterized by long and dilated times for effective innovation; hence, the fungal plague was always combated with a poor arsenal and limited suitable targets. Only in recent years have fungal infections been figured out as one of the most serious health problems of our time and for the near future, but the risks that they represent are not even remotely comparable to the number of available therapeutic options. Currently, a few classes of drugs are used in clinics and veterinary.
Polyenes
Amphotericin B is the major representative of the polyene class. It is a fungicide able to interfere with the ion homeostasis of the fungal cell, establishing hydrophobic interactions with ergosterol of the membrane and creating pores. It also induces an accumulation of reactive oxygen species and both mechanisms contribute to cell death. Since its discovery in 1955, more than 200 polyenes have been developed, but amphotericin B keeps being the drug of choice especially for systemic mycoses (Houšť et al. 2020). Although its high efficiency, its main limitations are represented by nephro- and hepatotoxicity, infusion reactions, and electrolyte anomalies (Laniado-Laborín and Cabrales-Vargas 2009; Nett and Andes 2016); therefore, from the deoxycholate form, new formulations were elaborated in order to ameliorate both safety and effectiveness. The most well-known are the lipid-based formulations that consist of the drug loading into liposomes or lipid complex, a colloidal dispersion and microemulsions (Marena et al. 2022).
Azoles
After their discovery, rapid azoles became the first-line drugs. Apart from a few exceptions, they are considered organism-dependent fungistatic/fungicidal (Manavathu et al. 1998) and can inhibit the cytochrome P450-dependent lanosterol-14α-demethylase, an essential enzyme in the synthesis of ergosterol (Mast et al. 2013). This class contains both imidazole- and triazole-active group molecules since several modifications in the structure followed one another, and gave birth to different generations. First generation is occupied for instance by clotrimazole, ketoconazole, miconazole, butoconazole, econazole, bifonazole, terconazole, tioconazole, etc., which present the imidazole ring. The scarce water solubility was an obstacle as well as the limited oral bioavailability and side effects like gastrointestinal and endocrine alterations and hepatotoxicity. Even if their uses decreased over time, especially for invasive infections, they are still adopted in topical preparations. Second and third generations exhibit a triazole ring in the formula and underwent structural changes that progressively resulted in increased efficacy and hydrosolubility, extended spectra, better pharmacokinetics and pharmacodynamics, lower toxicity, and new possible formulations. Fluconazole and itraconazole stand in the second generation while voriconazole, posaconazole, isavuconazole, ravuconazole, and albaconazole in the third (Nett and Andes 2016). They are widely used as treatment, prophylactic, and pre-emptive both in medical and surgical fields (Allen et al. 2015). Despite them being well-tolerated, a few restrictions have been highlighted like the QT prolongation, the teratogenicity, and the drug–drug interactions of triazoles (Pursley et al. 1996; Nett and Andes 2016; Cook et al. 2017).
Echinocandins
Echinocandins represent one of the most recent classes of antifungal drugs starting its development in the early 2000s. They are semisynthetic lipopeptides with a structure resembling fermentation metabolites of different microorganisms (Houšť et al. 2020). Echinocandins inhibit in a non-competitive manner the UDP-glucose-β-1,3-D-glucan-β-3-D-glucosyltransferase also known as β-1,3-D-glucan synthase, an essential enzyme for the synthesis of β-1,3-glucans, fundamental components of the fungal cell wall. β-1,3-D-glucan synthase is a complex formed by two subunits, one regulatory, Rho1 (Ras homologous 1), and one catalytic, Fks (FK506 sensitivity); in particular, Fks is encoded by three genes, FKS1, FKS2, and FKS3. Echinocandins seem to target specifically the FKS1 gene (Loh and Ang 2020). Based on the fungus and also on the species and strain, echinocandins can be considered either fungistatic or fungicidal. Caspofungin, micafungin, and anidulafungin were the most successful compounds that reached the market. Their strong activity associated with a broad spectrum, good pharmacokinetics and pharmacodynamics, high safety, and few drug–drug interactions makes them suitable as first-line drugs, especially in nosocomial candidemia (Kato et al. 2021).
Pyrimidine analogs
5-Flucytosine is the only component of this class. From its approval in the 1960s, it was considered for a long time just an antimetabolite for cancer treatment, but its low antineoplastic activity pushed it toward the antimicrobial pipeline as an antifungal drug. 5-Flucytosine is a fluorinated analog of cytosine, and for this reason, it can easily enter the fungal cells through the cytosine permease and be deaminated by cytosine deaminase to 5-fluorouracil. Then 5-fluorouracil is converted into 5-fluorouridine triphosphate able to be incorporated in fungal RNA, hence, altering the aminoacylation of tRNA and inhibiting the protein synthesis. At the same time, 5-fluorouracil, when metabolized into 5-fluorodeoxyuridine monophosphate, inhibits the thymidylate synthase, an essential enzyme for DNA synthesis and nuclear division (Houšť et al. 2020). This double mechanism of action makes 5-flucytosine a potent drug, especially in candidiasis and cryptococcosis, but it can be used only in combination with other drugs like amphotericin B because of the rapid development of resistance (Chandra et al. 2009). It has high oral bioavailability but a brief half-life. The interactions with other drugs are few but the main limitations are referred to bone marrow suppression and hepatotoxicity (Nett and Andes 2016).
Allylamines
Allylamines represent a small family of molecules that target the squalene epoxidase (or squalene monooxygenase) an enzyme needed for the ergosterol synthesis (Mota Fernandes et al. 2021). They are not commonly used in clinics except for dermatophytes against whom they are fungicidal (Ward et al. 2022). Terbinafine, naftifine and also pentamidine, and some derivatives like butenafine or amorolfine are examples of squalene epoxidase inhibitors.
The new
Some new strategies were found in recent years. As safety is the most important issue in patient treatment, the reasonable logic for target identification relies on the differences between fungal and host cells. Herein are reported molecules with antifungal properties acting on pathways presenting significant discrepancies between fungal and mammalian cells.
Hsp90-calcineurin-PKC axis
Among the relatively new targets, it is important to mention the calcineurin pathway (Fig. 1). Calcineurin is a Ca2+/calmodulin-activated protein phosphatase 2B essential for activating stress responses inside the host, but it also plays a role in fungal growth, morphotype change, virulence, and resistance development (Zheng et al. 2018). In response to stimulation, the calcium channel CMC (Cch1-Mid1 channel) in the plasma membrane lets Ca2+ ions enter the cell and bind calmodulin. The activated Ca2+-calmodulin complex, in turn, binds the calcineurin (Cn) heterodimer (CnA and CnB) which can dephosphorylate the calcineurin-dependent transcription factor calcineurin-responsive zinc finger 1 (Crz1). Crz1 then translocates into the nucleus and activates the transcription of genes related to stress response, cell wall integrity, growth, and drug resistance. Calcineurin is finely modulated by immunophilins cyclophilin A (CyP A) and FK506-binding protein 12 (FKBP 12). These proteins are the target of immunosuppressors like tacrolimus (FK506) and cyclosporine A, mainly used to avoid rejection after transplantations (Mota Fernandes et al. 2021). They bind immunophilins forming drug-immunophilin inhibitory complexes which impair the interaction of calcineurin to its substrates. Recently they received large interest for their synergistic effect in combination with caspofungin and azoles against different fungal species, echinocandins- and azoles-resistant strains as well, having an inhibitory activity also on the biofilm formation (Uppuluri et al. 2008; Lamoth et al. 2013; Şen Kaya et al. 2021). Nevertheless, because of the serious consequences on the immune systems, different efforts were made to limit side effects while still maintaining the antifungal activity. APX879, the successor of tacrolimus, has high efficacy in in vivo cryptococcosis but low immunosuppression (Juvvadi et al. 2019). New calcineurin inhibitors were discovered. Among them, there are vasostatin-I which is a peptide derived from chromogranin A, its shorter fragments chromofungin and catestatin, which are active against yeasts and filamentous fungi (Aslam et al. 2012; Jati et al. 2023), and tamoxifen, an anticancer that is supposed to interfere in the calcineurin pathway and that synergizes with amphotericin B (Hai et al. 2019).

Overview of the structural organization of a model fungal cell with a focus on the mechanism and pathway sources of potential antifungal targets. The pathways reported are those occurring in the cytosol or linked to the plasma membrane and cell wall. Arrows show the deep and complex interconnection among pathways giving a sight of the consequences of the activity of a single drug among the interconnected players and a possible explanation of the interactions between drugs
Calcineurin’s effectiveness is often associated with one of its upstream regulators, the heat shock protein 90 (Hsp90), a chaperon involved in stress signaling pathways, virulence, and resistance (Mota Fernandes et al. 2021). Hsp90 regulates the morphology of fungi differently; its inhibitors induce filamentation in Candida spp. (Shapiro et al. 2012) and repress it in Aspergillus spp. with negative outcomes in germination and conidiation (Lamoth et al. 2012). Hsp90 modulators, in combination with amphotericin B, azoles, and echinocandins improved the antifungal activity, reduced the mortality rate, and impaired the evolution of resistance (Singh et al. 2009). Mycograb (efungumab) is a monoclonal antibody in a single-chain-fragment variable format. It binds selectively to Hsp90 in the middle region. This inhibits the interaction between the C-terminal and N-terminal regions and prevents the conformational changes needed to perform its activity as a chaperon. Mycograb demonstrated great efficiency in invasive candidiasis (Karwa and Wargo 2009) but never reached the market because of quality and safety issues. Geldanamycin and its derivatives, such as the semi-synthetic variant 17-AAG, are other Hsp90 inhibitors. Besides their antitumoral properties, they showed good antifungal effects. They bind the N-terminal ATP/ADP-binding domain inhibiting the ATPase activity and proved to be effective both alone and in combination. Unfortunately, they also showed hepatotoxicity (Gorska 2012; Liu et al. 2022) mainly related to the cross-inhibition of Hsp90 of the host. For this reason, more selective inhibitors like CMLD013075, are currently under investigation (Whitesell et al. 2019). Other interesting Hsp90-targeting compounds include Ganetespib, able to reverse fluconazole resistance in an in vivo candidemia model (Yuan et al. 2021), and Monorden, suitable for the development of fungicides (Nguyen et al. 2020). Hsp90 is also involved in the maturation and stability of mTOR (mammalian target of rapamycin) effector complexes such as phosphatidylinositol 3-kinase-related protein kinases (PIKKs) (Takai et al. 2010; Millson and Piper 2014). The immunosuppressant rapamycin was demonstrated creating a complex with immunophilin FKBP 12. The complex then binds to the FRB (FKBP12-rapamycin binding) domain, in Tor protein kinases, inhibiting their activity. Tor protein kinases are fundamental components of TORC1 (target of rapamycin complex 1) which regulates a multitude of pathways (especially those nutrient-sensitive). When TOR protein kinases are inhibited by rapamycin, the fungal growth is blocked, and the azole resistance is reversed. Initially, its activity was supposed to be related just to the inhibition of TORC1 that regulates the gene expression of heat shock factor 1 (Hsf1) and, downstream, heat shock proteins like Hsp90 (Deprez et al. 2018). However, recently, rapamycin was observed to exert its antifungal effect also directly on Hsp90 by inhibiting Hsf1 activation without altering TORC1 (Millson and Piper 2014). Its main limitation is the immunosuppressive activity in the host, but some derivatives like everolimus, with reduced drawbacks, are under investigation also in combinatorial treatments (Jiang et al. 2022).
Hsp90 also regulates the signaling of the protein kinase C (PKC) cascade (Fig. 1) essential for calcineurin activation, cell wall integrity, growth, morphotype, resistance, and stress responses (LaFayette et al. 2010). Pkc1 is the key kinase of this cascade. It is activated by Rho1 and controls several pathways, including the MAPK (mitogen-activated protein kinase) cascade and the downstream Mkc1 (MAP kinase from C. albicans), Cek1 (C. albicans ERK-like kinase), and Hog1 (high osmolarity glycerol) signaling. The selective inhibition of Pkc1 by Cercosporamide reduced the virulence in vivo and conferred hypersensitivity to several drugs turning some fungistatic ones into fungicidal ones (Sussman et al. 2004). Moreover, the impaired activation of MAPK-Mkc1 decreased the resistance to ergosterol biosynthesis inhibitors opening the doors to potential upstream agents able to deplete Mkc1 or, in a similar way, Cek1 and Hog1 (Singh et al. 2009; LaFayette et al. 2010). In this regard, magnolol negatively affects C. albicans virulence factors by inhibiting PKC and MAPK-Cek1 signaling (Xie et al. 2022). Finally, another option concerns the inhibition of chaperons of the heat-shock protein family which are involved in the cascades. In particular, Hsp90 (see above) directly regulates Mkc1, Cek1, Hog1, and also Hsp70 (for Pkc1), which in turn are both regulated by Hsf1, Hsp21 (for Cek1), and Hsp12 (for Hog1).
At the top of the Hsp90-calcineurin-PKC axis, the histone deacetylases cascade (HDACs, KDACs) (Fig. 1) controls the expression and function of different proteins (Hsp90 included) sometimes involved in fungal virulence. AR-12 is a celecoxib analog protein kinase inhibitor that operates on different targets. It inhibits acetyl fungal coenzyme A synthetase 1 (Acetyl-CoA synthetase 1) causing a downstream repercussion on the metabolism and especially on the histone acetylation. Moreover, it affects Hsp90 and Hsp27 chaperons’ production, downregulating their genes (Rauseo et al. 2020). Noteworthy is Hos2 (HDA one similar), a regulator of Hsp90 deacetylation and activity. Among its inhibitors, there is MGCD290 which is extremely efficient against Candida and Aspergillus spp., especially when synergically combined with azoles and echinocandins (Rauseo et al. 2020). Trichostatin A is another HDAC inhibitor that enhanced the efficacy of azoles against C. albicans and of caspofungin against Aspergillus spp. (Bauer et al. 2016). Other interesting enzymes work at this level; when Hst3 is inhibited by nicotinamide, it demonstrates antifungal properties while genetic depletion of acetyltransferases conferred hypersensitivity to fluconazole (Wurtele et al. 2010).
Essentially inhibitors of the Hsp90-calcineurin-PKC axis represent actors that play at different but interconnected layers of the same piece. All the approaches are extremely promising in treating fungal infections.
The Ras pathway
Some other pathways are significantly different in fungi and in host cells; therefore, enzymes involved or genes encoding them could be optimal antifungal targets. This is the case of the Ras pathway (Fig. 1) in fungi that is involved in some protein transduction, virulence, morphotype switch, and vital processes (Fortwendel et al. 2004; Hogan and Sundstrom 2009). Ras proteins reach the plasma membrane both in a palmitoylated and not-palmitoylated form. There, some post-translational modifications essential for its biological activity occur, and finally, the interactions with the guanine nucleotide exchange factors (GEFs) like Cdc25 (cell division cycle 25) lead to activation. Brilliant antifungal approaches could exploit the inhibition of palmitoylation or of post-translational modifications such as prenylation. Alternatively, the impairment of the interaction of Ras with GEFs, with its common substrates, or with Ras-related proteins like RhbA are equally valuable strategies (Maurer et al. 2012; Wang et al. 2022). Some farnesyltransferase inhibitors proved to be successful in killing different fungal species (Hast et al. 2011; Qiao et al. 2013). For instance, tipifarnib and lonafarnib reversibly bind to the farnesyltransferase impeding the prenylation of the cysteine residue on the C-terminus of Ras protein (CAAX tetrapeptide motif), preventing its interaction with the plasma membrane and subsequent activation. Other farnesyltransferase inhibitors developed or discovered are farnesyltransferase inhibitor III which is able to block the yeast-hyphal change in C. albicans (McGeady et al. 2002), manumycin A, actinoplanic acid A, and its desmethyl analog actinoplanic acid B, isolated from, respectively, Streptomyces spp., Actinoplanes spp., and both of them (Mrak et al. 2018).
Lipids biosynthesis and lipid anchors
A similar discourse can be done for sphingolipids biosynthesis (Fig. 2). Even if they are components of the eukaryotic membranes, sphingolipids like inositol phosphoryl ceramide and glucosylceramide are responsible for virulence and drug resistance (Rittershaus et al. 2006; Fernandes et al. 2018). From the top of the sphingolipids biosynthesis, myriocin inhibits serine palmitoyl transferase (SPT), an enzyme that condensates L-serine and palmitoyl-Coenzyme A to produce 3-ketodihydrosphingosine (Yang et al. 2021). 3-ketodihydrosphingosine is reduced through 3-ketodihydrosphingosine reductase (KDSR) in dihydrosphingosine and acylated by ceramide synthase (CerS). Fumonisin B (Kozel and Wickes 2014) and australifungin act on the ceramide synthase whose impairment makes C. neoformans virulent (Delgado et al. 2006). Acylhydrazones like BHBM, D0, and derivatives like D2, D13, D17, and SB-AF-1002 affect the glucosylceramide synthesis by regulating the vesicle sorting or transport between Golgi apparatus and ER (Artunduaga Bonilla et al. 2021). In particular, BHBM and D0 affect the expression of APL5, COS111, MKK1, and STE2 genes which, in fact, are directly involved in vesicular trafficking of sphingolipids and cell cycle progression. Finally, aureobasidin A and its derivatives inhibit the inositol phosphoryl ceramide synthase (IPCS) which normally leads to the production of inositol phosphoryl ceramide (Aeed et al. 2009). In addition, there are also biological options. A IgG1, IgG2b, and a camelid single-domain antibody (VHH 41D01) showed their affinity for glucosylceramides and demonstrated high efficiency in inhibiting fungal growth. Moreover, the IgG1 offered protection from a lethal dose of C. neoformans; the IgG2b enhanced the macrophagic response, and the VHH 41D01 reduced the host inflammation (De Coninck et al. 2017). The recombinant enzyme Cerezyme can hydrolyze fungal glucosylceramides controlling C. neoformans infection and having a protective effect (Rhome et al. 2011).

Biosynthetic pathways occurring between the endoplasmic reticulum and the Golgi apparatus which are sources of antifungal targets. Some enzymes are already target of different compounds (Spt14 subunit of the UDP glycosyltransferase which catalyzes the formation of GlcNAc, Gwt1, and Mcd4 for the GPI-anchored protein (GPI AP) biosynthesis and SPT, CerS, and IPCS for the sphingolipids biosynthesis), but others are reported as potential new targets
Concerning post-translational modifications, some proteins are supplemented with a glycosylphosphatidylinositol (GPI) group that allows proteins to travel from the endoplasmic reticulum to the plasma membrane and to the cell wall (Fig. 2). An altered biosynthesis of the GPI anchor resulted in defective morphology and attenuated virulence both in C. albicans and A. fumigatus, meaning that some GPI-anchored proteins have a role in the fungal pathogenesis (Richard et al. 2002; Samalova et al. 2020; Snelders et al. 2022). The GPI-anchor biosynthesis became thereby a source of targets. GPI assembly initiates with the transfer of N-acetylglucosamine (GlcNAc) from UDP-GlcNAc to phosphatidylinositol (PI). This reaction is catalyzed by UDP-glycosyltransferase, and one of its subunits, Spt14 (suppressor of Ty insertion mutations), is the target of Jawsamycin. In the GPI-anchor biosynthesis, inositol acyltransferase Gwt1 (GPI-anchored wall protein transfer 1) mediates the acylation of glucosamine phosphatidylinositol (GlcN-PI) to form GlcN-(Acyl)-PI. Several compounds were found to inhibit Gwt1 like gepinacin, manogepix (APX001A or E1210), and its prodrug fosmanogepix (APX001 or E1211), BIQ, G365, and G884. Another interesting target is the phosphoethanolamine transferase-I Mcd4 (morphogenesis checkpoint dependent 4). Mcd4 transfers an ethanolamine phosphate (EtNP) group to the di-mannosylated GlcN-(Acyl)-PI (Man-Man-GlcN-(Acyl)-PI) to produce Man-(EtNP)-Man-GlcN-(Acyl)-PI. Molecules like YW3548 (M743) and M720 have Mcd4 as targets (Yadav and Khan 2018).
The trehalose pathway
One of the most attractive and newest targets is represented by trehalose biosynthesis (Fig. 1). Its absence in mammalian cells dramatically reduces the host toxicity. Trehalose is a storage carbohydrate useful as a carbon source for processes like glycolysis, fungal germination, and sporulation and is an important player in stress conditions. Its pathway involves two main enzymes: the trehalose 6-phosphate synthase 1 (Tps1) and the trehalose 6-phosphate phosphatase (Tps2). The deletion of their genes affected Cryptococcus spp., C. albicans, and Aspergillus spp. growth, survival, and virulence and Aspergillus spp. germination. Moreover, a compromised Tps2 causes an accumulation of trehalose 6-phosphate leading to cell death (Thammahong et al. 2017; Chen et al. 2020). Trehalose is used in fungal metabolism and for this reason, it is converted again into glucose thanks to trehalases (trehalose-degrading enzymes) which are α-glucosidase hydrolase. Validamycin A binds competitively with trehalase inhibiting its activity, thereby affecting the glucose metabolism of fungi. Furthermore, Validamycin seems to affect also genes related to MAPK and their ribosome synthesis. Despite these delicious targets, just a few studies have identified effective inhibitors (Yang et al. 2023).
The glyoxylate cycle
The glyoxylate cycle (Fig. 3) seems to be important for virulence and survival, especially after macrophagic engulfment. As for the trehalose pathway, some components involved in the glyoxylate cycle are completely absent in mammalian cells—isocitrate lyase (ICL) and malate synthase (MS) (Chen et al. 2020). Mohangamide A and mohangamide B are efficient inhibitors of ICL in C. albicans (Cheah et al. 2014); argentilactone and its semi-synthetic analogs inhibited ICL in Paracoccidioides, and some other compounds targeting MS are still under investigation (Prado et al. 2014).

Biosynthetic pathways and metabolic cycles occurring in the mitochondrion (the tricarboxylic acid cycle and the mitochondrial respiration), in the peroxisome (the glyoxylate cycle) or strictly linked to the mitochondrial structures (Pyrimidine biosynthesis). Herein are reported enzymes that are already targeted by different compounds (DHODH, the respiratory complexes, ICL, and MS) and potential new targets
Orotomides
With the term orotomides, the microbiological world has identified a completely new class of antifungal drugs. Their target is represented by the dihydroorotate dehydrogenase (DHODH) which is involved in the biosynthesis of pyrimidines (Fig. 3). The depletion of pyrimidines leads to blocks in the DNA/RNA synthesis and in the lipid and carbohydrate precursor productions. Olorofilm F901318 is an excellent inhibitor that can be selective for fungal DHODH even if the enzyme was found also in mammals. The only drawback is the narrow spectrum that includes molds and dimorphic fungi but not yeasts (Singh et al. 2021).
The respiratory chain
Among the promising targets, some can be found in the mitochondria (Fig. 3). They are the organelles adhibited to energy production through oxidative phosphorylation and the tricarboxylic acid cycle. Even if some intermediates and enzymes are shared by both fungi and host cells, there are some fungus-specific proteins suitable for new antifungal compound actions (Chatre and Ricchetti 2014). ATI-2307 and ilicicolin H are examples of the application of this approach. The former is an arylamidine able to inhibit both the complex III and IV of the respiratory chain interfering inevitably in the mitochondrial membrane potential (Shibata et al. 2012), while the latter acts on the mitochondrial cytochrome bc1 reductase (Singh et al. 2012). Both were tested on Candida, Cryptococcus, and Aspergillus spp., showing great potential, efficacy, and selectivity (Singh et al. 2012). Some other known molecules have been found to have antifungal effects altering the mitochondrial function. For instance, artemisinin seems to lead to mitochondrial dysfunction by affecting the transcription factor pleiotropic drug resistance 1 (PDR1) which regulates the drug efflux pumps and the ergosterol biosynthesis but also downregulates the NADH dehydrogenase Ndi1 of the mitochondrial electron transport chain. Quercetin affects the oxidative stress operating on some electron membrane transport proteins; histatin 5 disrupts the ATP synthesis machinery (e Silva et al. 2020), and VT-1129 and VT-1161 are potent selective inhibitors of the fungal cytochrome P45051 (CYP51). In addition, several antibacterial drugs have demonstrated antifungal properties. Most of them inhibit protein synthesis by binding the ribosome subunits but it seems that the inhibition of particular mitochondrial proteins is extremely deleterious for fungi. Moreover, some antibiotics appear to interfere also with the induction of stress-response mitochondrial chaperones. A few examples are represented by linezolid, azithromycin, and minocycline (Zhang et al. 2021a).
The cell wall
Some new strategies, looking at the current antifungal mechanisms, are directed toward the cell wall components or toward the enzymes that produce them (Fig. 1). Nikkomycin Z, for instance, is a competitive inhibitor of the chitin synthase because of the structural similarities with the substrate uridine diphosphate (UDP)-N-acetyl glucosamine (Larwood 2020). It has additive and synergic relationships with azoles and echinocandins in aspergillosis, candidiasis, and coccidioidomycosis (Larwood 2020). Polymyxins like polymyxin B have the same mechanism as nikkomycins and inhibit directly chitin synthase while plagiochin E alters the expression of chitin synthase genes (CHS). Lectins have a great affinity for polysaccharides like mannose, glucose, and chitin which represent the backbone and the body of the fungal cell wall (Wu et al. 2022). Other examples are represented by the monoclonal antibody IgG2b 2G8 and the humanized versions in different formats, the IgG1 H5K1 (Dia-T51) and the single-chain-fragment variable hscFv (scFv-3T). Their antigens are selectively β-1,3-glucans of the fungal cell wall. The first proved its efficacy in candidiasis, aspergillosis, and cryptococcosis but with the limitation of its murine nature (Di Mambro et al. 2021); while the others have great potential against Candida spp., especially when combined with amphotericin B and echinocandins (Di Mambro et al. 2021; Di Mambro et al. 2022; Vanzolini et al. 2023). Rezafungin (SP3025, CD101, or Rezzayo) is a lipopeptide that enters the class of echinocandins, and that in March 2023 got approval in the USA to be used for the treatment of candidemia and invasive candidiasis. It inhibits the β-1,3-glucan synthase enzyme complex, essential for β-1,3-glucans synthesis. Ibrexafungerp (SCY-078 or MK-3118) is a triterpene glycoside and the only FDA-approved antifungal for vulvovaginal candidiasis. As rezafungin, ibrexafungerp inhibits β-1,3-glucan synthase (Sucher et al. 2022; Phillips et al. 2023; Syed 2023). All these ways have the advantage of damaging the wall, proper just of fungi without repercussions for the host.
Trivalent cations-related pathways
A different strategy in antimicrobial development addresses iron-related pathways (Fig. 1). This gave the inspiration also for the development of new antifungal drugs. Iron is hijacked by fungal cells and is needed for many processes that confer the typical virulence factors. The main objective is the impairment or the exploitation of the iron assimilation. Different strategies have been adopted. Some antifungal peptides were conjugated to the siderophores in order to be incorporated inside the cells where they are released to carry out their task (Tonziello et al. 2019). Other compounds interfere with the synthesis of siderophores by inhibiting enzymes like nonribosomal peptide synthetases (NRPS), polyketide synthase (PKS), phosphopantetheine transferase (PPtase), and nonribosomal peptide synthetases (NRPS)-independent siderophore synthetases (NIS). In this context, celastrol inhibits A. fumigatus growth by acting on the flavin-dependent monooxygenase siderophore A (SidA) (Sass et al. 2019). Other methods exploit iron chelators or the substitution of iron with gallium (Bastos et al. 2019). Lipocalins, especially ASP-2397, seem to chelate Al3+ and Fe3+ and to be translocated inside the fungal cell through the siderophore iron transporter 1 (Sit1), preventing the cations hijacking. It showed good selectivity because of the lack of the Sit1 in mammalian cells, and high potency against different fungi (Nakamura et al. 2019). Ciclopirox has a similar mechanism of action sequestering Al3+ and Fe3+ (Subissi et al. 2010; Oltu et al. 2020a). Salinomycin, deeply studied as an anticancer agent, presents a multimodal mechanism, but the one that appears to be related to the antifungal effect is represented by the complexation of metal cations and their transportation inside the cells by acting as carboxylic polyether ionophore. It is active against some yeasts and filamentous fungi, especially in combination with polymyxin B (Antoszczak and Huczyński 2019).
Other targets
Fungal mitosis is another interesting target and, among its inhibitors, the polyketide griseofulvin binds to α and β tubulin, thus inhibiting the formation of the mitotic tube, hence the mitosis and the nuclear acid synthesis (Aris et al. 2022). Haloprogin activity seems related to the inhibition of oxygen uptake and subsequent membrane disruption. It is probably associated with the block of glutathione activity that occurs also for allicin, the molecule of which haloprogin is the active part (Oltu et al. 2020b). With the same rationale, inhibitors of the superoxide dismutases, catalases, and peroxidases are tightly correlated to the remotion of ROS produced by the immune system cells making the fungal cell unable to protect itself (Oltu et al. 2020b). Some cyclooxygenase inhibitors showed antibiofilm activity by inhibiting the prostaglandin E2 biosynthesis (Houšť et al. 2020), and haemofungin has a potent inhibitory activity toward the HemH/ferrochelatase, which is an enzyme that catalyzes the conversion of highly phototoxic porphyrin molecules into haem.
Concluding remarks
Several natural, semi-synthetic, and synthetic molecules are under investigation as antifungal drugs or as enhancers for commercially available drugs (summarized in Table 1) (e Silva et al. 2020). Concurrently, different repurposing studies are arising to enrich the drug arsenal (Zhang et al. 2021a). No antifungal vaccines have been licensed yet, but research on them and on adjuvants is ongoing. Due to a multiplicity of obstacles, fungal infections keep being a big challenge (Oliveira et al. 2021). Regarding that, metabolomic, proteomic, and lipidomic sciences in combination with computational biology have granted great steps forward in the identification of new targets and compounds in faster times (Amarsaikhan et al. 2017; Brandt et al. 2020; Shahi et al. 2020). We hope that these advancements can also meet facilitated and accelerated paths for entry in the market of new efficient diagnostic and therapeutic tools.
References
Aeed PA, Young CL, Nagiec MM, Elhammer AP (2009) Inhibition of inositol phosphorylceramide synthase by the cyclic peptide aureobasidin A. Antimicrob Agents Chemother 53:496–504. https://doi.org/10.1128/AAC.00633-08
Allen D, Wilson D, Drew R, Perfect J (2015) Azole antifungals: 35 years of invasive fungal infection management. Expert Rev Anti Infect Ther 13:787–798. https://doi.org/10.1586/14787210.2015.1032939
Amarsaikhan N, Albrecht-Eckardt D, Sasse C, Braus GH, Ogel ZB, Kniemeyer O (2017) Proteomic profiling of the antifungal drug response of Aspergillus fumigatus to voriconazole. Int J Med Microbiol IJMM 307:398–408. https://doi.org/10.1016/j.ijmm.2017.07.011
Antoszczak M, Huczyński A (2019) Salinomycin and its derivatives – a new class of multiple-targeted “magic bullets”. Eur J Med Chem 176:208–227. https://doi.org/10.1016/j.ejmech.2019.05.031
Aris P, Wei Y, Mohamadzadeh M, Xia X (2022) Griseofulvin: an updated overview of old and current knowledge. Mol Basel Switz 27:7034. https://doi.org/10.3390/molecules27207034
Artunduaga Bonilla JJ, Honorato L, Haranahalli K, Dib Ferreira Gremião I, Pereira SA, Guimarães A, de Souza Baptista AR, de Melo Tavares P, Rodrigues ML, Miranda K, Ojima I (2021) Antifungal activity of acylhydrazone derivatives against Sporothrix spp. Antimicrob Agents Chemother 65:e02593-20, AAC.02593-20. https://doi.org/10.1128/AAC.02593-20
Aslam R, Atindehou M, Lavaux T, Haïkel Y, Schneider F, Metz-Boutigue M-H (2012) Chromogranin A-derived peptides are involved in innate immunity. Curr Med Chem 19:4115–4123. https://doi.org/10.2174/092986712802430063
Bastos RW, Rossato L, Valero C, Lagrou K, Colombo AL, Goldman GH (2019) Potential of gallium as an antifungal agent. Front Cell Infect Microbiol 9:414. https://doi.org/10.3389/fcimb.2019.00414
Bauer I, Varadarajan D, Pidroni A, Gross S, Vergeiner S, Faber B, Hermann M, Tribus M, Brosch G, Graessle S (2016) A class 1 histone deacetylase with potential as an antifungal target. mBio 7:e00831-16. https://doi.org/10.1128/mBio.00831-16
Brandt P, Garbe E, Vylkova S (2020) Catch the wave: metabolomic analyses in human pathogenic fungi. PLOS Pathog 16:e1008757. https://doi.org/10.1371/journal.ppat.1008757
Chandra J, Mohammad S, Ghannoum MA (2009) Flucytosine: site of action, mechanism of resistance and use in combination therapy. In: Mayers DL (ed) Antimicrobial drug resistance: mechanisms of drug resistance. Humana Press, Totowa, NJ, pp 313–326
Chatre L, Ricchetti M (2014) Are mitochondria the Achilles’ heel of the kingdom fungi? Curr Opin Microbiol 20:49–54. https://doi.org/10.1016/j.mib.2014.05.001
Cheah H-L, Lim V, Sandai D (2014) Inhibitors of the glyoxylate cycle enzyme ICL1 in Candida albicans for potential use as antifungal agents. PloS One 9:e95951. https://doi.org/10.1371/journal.pone.0095951
Chen X, Zhang Z, Chen Z, Li Y, Su S, Sun S (2020) Potential antifungal targets based on glucose metabolism pathways of Candida albicans. Front Microbiol 11:296. https://doi.org/10.3389/fmicb.2020.00296
Cook K, Sraubol T, Campbell KB, Mourad A, Stiber J, Perfect JR, Johnson M (2017) QTc Prolongation in patients receiving triazoles and amiodarone. Open Forum Infect Dis 4:S84. https://doi.org/10.1093/ofid/ofx163.031
De Coninck B, Verheesen P, Vos CM, Van Daele I, De Bolle MF, Vieira JV, Peferoen M, Cammue BPA, Thevissen K (2017) Fungal glucosylceramide-specific camelid single domain antibodies are characterized by broad spectrum antifungal activity. Front Microbiol 8:1059. https://doi.org/10.3389/fmicb.2017.01059
Delgado A, Casas J, Llebaria A, Abad JL, Fabrias G (2006) Inhibitors of sphingolipid metabolism enzymes. Biochim Biophys Acta BBA - Biomembr 1758:1957–1977. https://doi.org/10.1016/j.bbamem.2006.08.017
Deprez M-A, Eskes E, Winderickx J, Wilms T (2018) The TORC1-Sch9 pathway as a crucial mediator of chronological lifespan in the yeast Saccharomyces cerevisiae. FEMS Yeast Res 18. https://doi.org/10.1093/femsyr/foy048
Di Mambro T, Vanzolini T, Bianchi M, Crinelli R, Canonico B, Tasini F, Menotta M, Magnani M (2022) Development and in vitro characterization of a humanized scFv against fungal infections. PloS One 17:e0276786. https://doi.org/10.1371/journal.pone.0276786
Di Mambro T, Vanzolini T, Bruscolini P, Perez-Gaviro S, Marra E, Roscilli G, Bianchi M, Fraternale A, Schiavano GF, Canonico B, Magnani M (2021) A new humanized antibody is effective against pathogenic fungi in vitro. Sci Rep 11:19500. https://doi.org/10.1038/s41598-021-98659-5
e Silva KS, Silva L, Gonçales R, Neves B, Soares C, Pereira M (2020) Setting new routes for antifungal drug discovery against pathogenic fungi. Curr Pharm Des 26. https://doi.org/10.2174/1381612826666200317125956
Fernandes CM, Goldman GH, Del Poeta M (2018) Biological roles played by sphingolipids in dimorphic and filamentous fungi. mBio 9:e00642-18. https://doi.org/10.1128/mBio.00642-18
Fortwendel JR, Panepinto JC, Seitz AE, Askew DS, Rhodes JC (2004) Aspergillus fumigatus rasA and rasB regulate the timing and morphology of asexual development. Fungal Genet Biol 41:129–139. https://doi.org/10.1016/j.fgb.2003.10.004
Gorska M (2012) Geldanamycin and its derivatives as Hsp90 inhibitors. Front Biosci 17:2269. https://doi.org/10.2741/4050
Hai TP, Van AD, Ngan NTT, Nhat LTH, Lan NPH, Vinh Chau NV, Thwaites GE, Krysan D, Day JN (2019) The combination of tamoxifen with amphotericin B, but not with fluconazole, has synergistic activity against the majority of clinical isolates of Cryptococcus neoformans. Mycoses 62:818–825. https://doi.org/10.1111/myc.12955
Hast MA, Nichols CB, Armstrong SM, Kelly SM, Hellinga HW, Alspaugh JA, Beese LS (2011) Structures of Cryptococcus neoformans protein farnesyltransferase reveal strategies for developing inhibitors that target fungal pathogens. J Biol Chem 286:35149–35162. https://doi.org/10.1074/jbc.M111.250506
Hogan DA, Sundstrom P (2009) The Ras/cAMP/PKA signaling pathway and virulence in Candida albicans. Future Microbiol 4:1263–1270. https://doi.org/10.2217/fmb.09.106
Houšť J, Spížek J, Havlíček V (2020) Antifungal drugs. Metabolites 10:106. https://doi.org/10.3390/metabo10030106
Jati S, Mahata S, Das S, Chatterjee S, Mahata SK (2023) Catestatin: antimicrobial functions and potential therapeutics. Pharmaceutics 15:1550. https://doi.org/10.3390/pharmaceutics15051550
Jiang H, Xiong J, Tan L, Jin P, Sun Y, Yang L, Tan J (2022) In vitro interactions of antifungal agents and everolimus against Aspergillus species. Front Cell Infect Microbiol 12:936814. https://doi.org/10.3389/fcimb.2022.936814
Juvvadi PR, Fox D, Bobay BG, Hoy MJ, Gobeil SMC, Venters RA, Chang Z, Lin JJ, Averette AF, Cole DC, Barrington BC, Wheaton JD, Ciofani M, Trzoss M, Li X, Lee SC, Chen Y-L, Mutz M, Spicer LD et al (2019) Harnessing calcineurin-FK506-FKBP12 crystal structures from invasive fungal pathogens to develop antifungal agents. Nat Commun 10:4275. https://doi.org/10.1038/s41467-019-12199-1
Karwa R, Wargo KA (2009) Efungumab: a novel agent in the treatment of invasive candidiasis. Ann Pharmacother 43:1818–1823. https://doi.org/10.1345/aph.1M218
Kato H, Hagihara M, Shibata Y, Asai N, Yamagishi Y, Iwamoto T, Mikamo H (2021) Comparison of mortality between echinocandins and polyenes for an initial treatment of candidemia: a systematic review and meta-analysis. J Infect Chemother Off J Jpn Soc Chemother 27:1562–1570. https://doi.org/10.1016/j.jiac.2021.06.017
Kozel TR, Wickes B (2014) Fungal diagnostics. Cold Spring Harb Perspect Med 4:a019299. https://doi.org/10.1101/cshperspect.a019299
LaFayette SL, Collins C, Zaas AK, Schell WA, Betancourt-Quiroz M, Gunatilaka AAL, Perfect JR, Cowen LE (2010) PKC signaling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, Calcineurin, and Hsp90. PLOS Pathog 6:e1001069. https://doi.org/10.1371/journal.ppat.1001069
Lamoth F, Juvvadi PR, Fortwendel JR, Steinbach WJ (2012) Heat shock protein 90 is required for conidiation and cell wall integrity in Aspergillus fumigatus. Eukaryot Cell 11:1324–1332. https://doi.org/10.1128/EC.00032-12
Lamoth F, Juvvadi PR, Gehrke C, Steinbach WJ (2013) In vitro activity of calcineurin and heat shock protein 90 inhibitors against Aspergillus fumigatus azole- and echinocandin-resistant strains. Antimicrob Agents Chemother 57:1035–1039. https://doi.org/10.1128/AAC.01857-12
Laniado-Laborín R, Cabrales-Vargas MN (2009) Amphotericin B: side effects and toxicity. Rev Iberoam Micol 26:223–227. https://doi.org/10.1016/j.riam.2009.06.003
Larwood DJ (2020) Nikkomycin Z-ready to meet the promise? J Fungi Basel Switz 6:261. https://doi.org/10.3390/jof6040261
Liu L, Zhang X, Kayastha S, Tan L, Zhang H, Tan J, Li L, Mao J, Sun Y (2022) A preliminary in vitro and in vivo evaluation of the effect and action mechanism of 17-AAG combined with azoles against azole-resistant Candida spp. Front Microbiol 13:825745. https://doi.org/10.3389/fmicb.2022.825745
Loh BS, Ang WH (2020) “Illuminating” echinocandins mechanism of action. ACS Cent Sci 6:1651–1653. https://doi.org/10.1021/acscentsci.0c01222
Manavathu EK, Cutright JL, Chandrasekar PH (1998) Organism-dependent fungicidal activities of azoles. Antimicrob Agents Chemother 42:3018–3021
Marena GD, Ramos MADS, Bauab TM, Chorilli M (2022) A critical review of analytical methods for quantification of amphotericin B in biological samples and pharmaceutical formulations. Crit Rev Anal Chem 52:555–576. https://doi.org/10.1080/10408347.2020.1811947
Mast N, Zheng W, Stout CD, Pikuleva IA (2013) Antifungal azoles: structural insights into undesired tight binding to cholesterol-metabolizing CYP46A1. Mol Pharmacol 84:86–94. https://doi.org/10.1124/mol.113.085902
Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, Fauber BP, Pan B, Malek S, Stokoe D, Ludlam MJC, Bowman KK, Wu J, Giannetti AM, Starovasnik MA, Mellman I, Jackson PK, Rudolph J, Wang W, Fang G (2012) Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci USA 109:5299–5304. https://doi.org/10.1073/pnas.1116510109
McGeady P, Logan DA, Wansley DL (2002) A protein-farnesyl transferase inhibitor interferes with the serum-induced conversion of Candida albicans from a cellular yeast form to a filamentous form. FEMS Microbiol Lett 213:41–44. https://doi.org/10.1111/j.1574-6968.2002.tb11283.x
Millson SH, Piper PW (2014) Insights from yeast into whether the rapamycin inhibition of heat shock transcription factor (Hsf1) can prevent the Hsf1 activation that results from treatment with an Hsp90 inhibitor. Oncotarget 5:5054–5064
Mota Fernandes C, Dasilva D, Haranahalli K, McCarthy JB, Mallamo J, Ojima I, Del Poeta M (2021) The future of antifungal drug therapy: novel compounds and targets. Antimicrob Agents Chemother 65:e01719–e01720. https://doi.org/10.1128/AAC.01719-20
Mrak P, Krastel P, Pivk Lukančič P, Tao J, Pistorius D, Moore CM (2018) Discovery of the actinoplanic acid pathway in Streptomyces rapamycinicus reveals a genetically conserved synergism with rapamycin. J Biol Chem 293:19982–19995. https://doi.org/10.1074/jbc.RA118.005314
Nakamura I, Ohsumi K, Takeda S, Katsumata K, Matsumoto S, Akamatsu S, Mitori H, Nakai T (2019) ASP2397 Is a novel natural compound that exhibits rapid and potent fungicidal activity against Aspergillus species through a specific transporter. Antimicrob Agents Chemother 63:e02689–e02618. https://doi.org/10.1128/AAC.02689-18
Nett JE, Andes DR (2016) Antifungal agents: spectrum of activity, pharmacology, and clinical indications. Infect Dis Clin North Am 30:51–83. https://doi.org/10.1016/j.idc.2015.10.012
Nguyen HTT, Choi S, Kim S, Lee J-H, Park AR, Yu NH, Yoon H, Bae C-H, Yeo JH, Choi GJ, Son H, Kim J-C (2020) The Hsp90 inhibitor, monorden, is a promising lead compound for the development of novel fungicides. Front Plant Sci 11:371. https://doi.org/10.3389/fpls.2020.00371
Oliveira LVN, Wang R, Specht CA, Levitz SM (2021) Vaccines for human fungal diseases: close but still a long way to go. Npj Vaccines 6:1–8. https://doi.org/10.1038/s41541-021-00294-8
Oltu I, Cepoi L, Rudic V, Rudi L, Chiriac T, Valuta A, Codreanu S (2020a) Current research and new perspectives in antifungal drug development. Adv Exp Med Biol 1282:71–83. https://doi.org/10.1007/5584_2019_453
Oltu I, Cepoi L, Rudic V, Rudi L, Chiriac T, Valuta A, Codreanu S (2020b) Current research and new perspectives in antifungal drug development. In: Donelli G (ed) Advances in microbiology, infectious diseases and public health: Volume 14. Springer International Publishing, Cham, pp 71–83
Phillips NA, Rocktashel M, Merjanian L (2023) Ibrexafungerp for the treatment of vulvovaginal candidiasis: design, development and place in therapy. Drug Des Devel Ther 17:363–367. https://doi.org/10.2147/DDDT.S339349
Prado RSD, Alves RJ, Oliveira CMAD, Kato L, Silva RAD, Quintino GO, do Desterro Cunha S, de Almeida Soares CM, Pereira M (2014) Inhibition of Paracoccidioides lutzii Pb01 isocitrate lyase by the natural compound argentilactone and its semi-synthetic derivatives. PLOS ONE 9:e94832. https://doi.org/10.1371/journal.pone.0094832
Pursley TJ, Blomquist IK, Abraham J, Andersen HF, Bartley JA (1996) Fluconazole-induced congenital anomalies in three infants. Clin Infect Dis Off Publ Infect Dis Soc Am 22:336–340. https://doi.org/10.1093/clinids/22.2.336
Qiao J, Gao P, Jiang X, Fang H (2013) In vitro antifungal activity of farnesyltransferase inhibitors against clinical isolates of Aspergillus and Candida. Ann Clin Microbiol Antimicrob 12:37. https://doi.org/10.1186/1476-0711-12-37
Rauseo AM, Coler-Reilly A, Larson L, Spec A (2020) Hope on the horizon: novel fungal treatments in development. Open Forum. Infect Dis 7:ofaa016. https://doi.org/10.1093/ofid/ofaa016
Rhome R, Singh A, Kechichian T, Drago M, Morace G, Luberto C, Poeta MD (2011) Surface localization of glucosylceramide during Cryptococcus neoformans infection allows targeting as a potential antifungal. PLOS ONE 6:e15572. https://doi.org/10.1371/journal.pone.0015572
Richard M, Ibata-Ombetta S, Dromer F, Bordon-Pallier F, Jouault T, Gaillardin C (2002) Complete glycosylphosphatidylinositol anchors are required in Candida albicans for full morphogenesis, virulence and resistance to macrophages. Mol Microbiol 44:841–853. https://doi.org/10.1046/j.1365-2958.2002.02926.x
Rittershaus PC, Kechichian TB, Allegood JC, Merrill AH, Hennig M, Luberto C, Poeta MD (2006) Glucosylceramide synthase is an essential regulator of pathogenicity of Cryptococcus neoformans. J Clin Invest 116:1651–1659. https://doi.org/10.1172/JCI27890
Rodrigues ML, Nosanchuk JD (2020) Fungal diseases as neglected pathogens: a wake-up call to public health officials. PLoS Negl Trop Dis 14:e0007964. https://doi.org/10.1371/journal.pntd.0007964
Samalova M, Carr P, Bromley M, Blatzer M, Moya-Nilges M, Latgé J-P, Mouyna I (2020) GPI Anchored proteins in Aspergillus fumigatus and cell wall morphogenesis. Curr Top Microbiol Immunol 425:167–186. https://doi.org/10.1007/82_2020_207
Sass G, Ansari SR, Dietl A-M, Déziel E, Haas H, Stevens DA (2019) Intermicrobial interaction: Aspergillus fumigatus siderophores protect against competition by Pseudomonas aeruginosa. PLOS ONE 14:e0216085. https://doi.org/10.1371/journal.pone.0216085
Şen Kaya S, Kiraz N, Bariş A, Turan D, Öz Y, Dağ İ, Aygün G (2021) Effects of calcineurin inhibitors, cyclosporine A and tacrolimus (FK506), on the activity of antifungal drugs against Candida spp. J Med Microbiol 70. https://doi.org/10.1099/jmm.0.001354
Shahi G, Kumar M, Kumari S, Rudramurthy SM, Chakrabarti A, Gaur NA, Singh A, Prasad R (2020) A detailed lipidomic study of human pathogenic fungi Candida auris. FEMS Yeast Res 20:foaa045. https://doi.org/10.1093/femsyr/foaa045
Shapiro RS, Sellam A, Tebbji F, Whiteway M, Nantel A, Cowen LE (2012) Pho85, Pcl1, and Hms1 Signaling governs Candida albicans morphogenesis induced by high temperature or Hsp90 compromise. Curr Biol 22:461–470. https://doi.org/10.1016/j.cub.2012.01.062
Shibata T, Takahashi T, Yamada E, Kimura A, Nishikawa H, Hayakawa H, Nomura N, Mitsuyama J (2012) T-2307 causes collapse of mitochondrial membrane potential in yeast. Antimicrob Agents Chemother 56:5892–5897. https://doi.org/10.1128/AAC.05954-11
Singh A, Singh P, Meis JF, Chowdhary A (2021) In vitro activity of the novel antifungal olorofim against dermatophytes and opportunistic moulds including Penicillium and Talaromyces species. J Antimicrob Chemother 76:1229–1233. https://doi.org/10.1093/jac/dkaa562
Singh SB, Liu W, Li X, Chen T, Shafiee A, Card D, Abruzzo G, Flattery A, Gill C, Thompson JR, Rosenbach M, Dreikorn S, Hornak V, Meinz M, Kurtz M, Kelly R, Onishi JC (2012) Antifungal spectrum, in vivo efficacy, and structure–activity relationship of ilicicolin H. ACS Med Chem Lett 3:814–817. https://doi.org/10.1021/ml300173e
Singh SD, Robbins N, Zaas AK, Schell WA, Perfect JR, Cowen LE (2009) Hsp90 Governs echinocandin resistance in the pathogenic yeast Candida albicans via Calcineurin. PLOS Pathog 5:e1000532. https://doi.org/10.1371/journal.ppat.1000532
Snelders E, Moyrand F, Sturny-Leclère A, Vernel-Pauillac F, Volant S, Janbon G, Alanio A (2022) The role of glycosylphosphatidylinositol (gpi) anchored proteins in Cryptococcus neoformans. Microbes Infect 24:105016. https://doi.org/10.1016/j.micinf.2022.105016
Subissi A, Monti D, Togni G, Mailland F (2010) Ciclopirox: recent nonclinical and clinical data relevant to its use as a topical antimycotic agent. Drugs 70:2133–2152. https://doi.org/10.2165/11538110-000000000-00000
Sucher AJ, Thai A, Tran C, Mantena N, Noronha A, Chahine EB (2022) Ibrexafungerp: a new triterpenoid antifungal. Am J Health-Syst Pharm 79:2208–2221. https://doi.org/10.1093/ajhp/zxac256
Sussman A, Huss K, Chio L-C, Heidler S, Shaw M, Ma D, Zhu G, Campbell RM, Park T-S, Kulanthaivel P, Scott JE, Carpenter JW, Strege MA, Belvo MD, Swartling JR, Fischl A, Yeh W-K, Shih C, Ye XS (2004) Discovery of cercosporamide, a known antifungal natural product, as a selective Pkc1 kinase inhibitor through high-throughput screening. Eukaryot cell 3:932–943. https://doi.org/10.1128/EC.3.4.932-943.2004
Syed YY (2023) Rezafungin: first approval. Drugs 83:833–840. https://doi.org/10.1007/s40265-023-01891-8
Takai H, Xie Y, de Lange T, Pavletich NP (2010) Tel2 structure and function in the Hsp90-dependent maturation of mTOR and ATR complexes. Genes Dev 24:2019–2030. https://doi.org/10.1101/gad.1956410
Thammahong A, Puttikamonkul S, Perfect JR, Brennan RG, Cramer RA (2017) Central role of the trehalose biosynthesis pathway in the pathogenesis of human fungal infections: opportunities and challenges for therapeutic development. Microbiol Mol Biol Rev MMBR 81:e00053–e00016. https://doi.org/10.1128/MMBR.00053-16
Tonziello G, Caraffa E, Pinchera B, Granata G, Petrosillo N (2019) Present and future of siderophore-based therapeutic and diagnostic approaches in infectious diseases. Infect Dis Rep 11. https://doi.org/10.4081/idr.2019.8208
Uppuluri P, Nett J, Heitman J, Andes D (2008) Synergistic effect of calcineurin inhibitors and fluconazole against Candida albicans biofilms. Antimicrob Agents Chemother 52:1127–1132. https://doi.org/10.1128/AAC.01397-07
Vanzolini T, Mambro TD, Magnani M, Menotta M (2023) AFM evaluation of a humanized recombinant antibody affecting C. auris cell wall and stability. RSC Adv 13:6130–6142. https://doi.org/10.1039/D2RA07217C
Wang Y, Xu F, Nichols CB, Shi Y, Hellinga HW, Alspaugh JA, Distefano MD, Beese LS (2022) Structure-guided discovery of potent antifungals that prevent Ras signaling by inhibiting protein farnesyltransferase. J Med Chem 65:13753–13770. https://doi.org/10.1021/acs.jmedchem.2c00902
Ward H, Parkes N, Smith C, Kluzek S, Pearson R (2022) Consensus for the treatment of tinea pedis: a systematic review of randomised controlled trials. J Fungi 8:351. https://doi.org/10.3390/jof8040351
Whitesell L, Robbins N, Huang DS, McLellan CA, Shekhar-Guturja T, LeBlanc EV, Nation CS, Hui R, Hutchinson A, Collins C, Chatterjee S, Trilles R, Xie JL, Krysan DJ, Lindquist S, Porco JA, Tatu U, Brown LE, Pizarro J, Cowen LE (2019) Structural basis for species-selective targeting of Hsp90 in a pathogenic fungus. Nat Commun 10:402. https://doi.org/10.1038/s41467-018-08248-w
Wickes BL, Wiederhold NP (2018) Molecular diagnostics in medical mycology. Nat Commun 9:5135. https://doi.org/10.1038/s41467-018-07556-5
Wu Y, Zhang M, Yang Y, Ding X, Yang P, Huang K, Hu X, Zhang M, Liu X, Yu H (2022) Structures and mechanism of chitin synthase and its inhibition by antifungal drug Nikkomycin Z. Cell Discov 8:129. https://doi.org/10.1038/s41421-022-00495-y
Wurtele H, Tsao S, Lépine G, Mullick A, Tremblay J, Drogaris P, Lee E-H, Thibault P, Verreault A, Raymond M (2010) Modulation of histone H3 lysine 56 acetylation as an antifungal therapeutic strategy. Nat Med 16:774–780. https://doi.org/10.1038/nm.2175
Xie Y, Hua H, Zhou P (2022) Magnolol as a potent antifungal agent inhibits Candida albicans virulence factors via the PKC and Cek1 MAPK signaling pathways. Front Cell Infect Microbiol 12:935322. https://doi.org/10.3389/fcimb.2022.935322
Yadav U, Khan MA (2018) Targeting the GPI biosynthetic pathway. Pathog Glob Health 112:115–122. https://doi.org/10.1080/20477724.2018.1442764
Yang X, Pei Z, Hu R, Zhang Z, Lou Z, Sun X (2021) Study on the inhibitory activity and possible mechanism of myriocin on clinically relevant drug-resistant Candida albicans and its biofilms. Biol Pharm Bull 44:305–315. https://doi.org/10.1248/bpb.b20-00246
Yang X, Shu Y, Cao S, Sun H, Zhang X, Zhang A, Li Y, Ma D, Chen H, Li W (2023) Trehalase inhibitor validamycin may have additional mechanisms of toxicology against Rhizoctonia cerealis. J Fungi 9:846. https://doi.org/10.3390/jof9080846
Yuan R, Tu J, Sheng C, Chen X, Liu N (2021) Effects of Hsp90 inhibitor ganetespib on inhibition of azole-resistant Candida albicans. Front Microbiol 12:680382. https://doi.org/10.3389/fmicb.2021.680382
Zhang Q, Liu F, Zeng M, Mao Y, Song Z (2021a) Drug repurposing strategies in the development of potential antifungal agents. Appl Microbiol Biotechnol 1–21. https://doi.org/10.1007/s00253-021-11407-7
Zhang SX, Babady NE, Hanson KE, Harrington AT, PMK L, Leal SM, Luethy PM, Martin IW, Pancholi P, Procop GW, Riedel S, Seyedmousavi S, Sullivan KV, Walsh TJ, Lockhart SR, Fungal Diagnostics Laboratories Consortium (FDLC) (2021b) Recognition of diagnostic gaps for laboratory diagnosis of fungal diseases: expert opinion from the Fungal Diagnostics Laboratories Consortium (FDLC). J Clin Microbiol 59:e0178420. https://doi.org/10.1128/JCM.01784-20
Zheng Y-H, Ma Y-Y, Ding Y, Chen X-Q, Gao G-X (2018) An insight into new strategies to combat antifungal drug resistance. Drug Des Devel Ther 12:3807–3816. https://doi.org/10.2147/DDDT.S185833
Acknowledgements
We would like to acknowledge the contribution of Tomas Di Mambro Ph.D. and Professor Francesco Barchiesi for having coauthored and significantly contributed among others in the papers cited in this manuscript and, in general, to the anti/fungal knowledge. The figures were partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
Funding
Open access funding provided by Università degli Studi di Urbino Carlo Bo within the CRUI-CARE Agreement. This work has been funded by the European Union—NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041—VITALITY—CUP H33C22000430006.
Author information
Authors and Affiliations
Contributions
T.V. carried out the literature study and drafted the manuscript. M.M. edited and reviewed. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This article does not contain any studies with human participants performed by any of the authors.
Competing interests
Diatheva s.r.l. supported some studies cited in the text. M.M. holds shares in Diatheva s.r.l. This does not alter our adherence to the Journal policies. T.V. declares that she has no financial interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vanzolini, T., Magnani, M. Old and new strategies in therapy and diagnosis against fungal infections. Appl Microbiol Biotechnol 108, 147 (2024). https://doi.org/10.1007/s00253-023-12884-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00253-023-12884-8