
Abstract
The biocatalytic generation of high-value chemicals from abundant, cheap and renewable feedstocks is an area of great contemporary interest. A strain of Rhodococcus erythropolis designated MLT1 was isolated by selective enrichment from the soil surrounding hop plants, using the abundant triene β-myrcene from hops as a sole carbon source for growth. Resting cells of the organism were challenged with β-myrcene, and the major product of biotransformation was determined by mass spectrometric analysis to be the monoterpene alcohol geraniol. Controls demonstrated that the product was biogenic and that an aerobic environment was required. The ability to transform β-myrcene was shown to be restricted to cells that had been grown on this substrate as sole carbon source. Pre-incubation of cells with the cytochrome P450 inhibitors metyrapone or 1-aminobenzotriazole reduced geraniol production by 23% and 73% respectively, but reduction in activity was found not to correlate with the inhibitor concentration. A comparative analysis of insoluble and soluble cell extracts derived from cells of MLT1 grown on either β-myrcene or glucose revealed at least four proteins that were clearly overproduced in response to growth on β-myrcene. Mass spectrometric analysis of tryptic digests of three of these protein bands suggested their identities as an aldehyde dehydrogenase, an acyl-CoA dehydrogenase and a chaperone-like protein, each of which has a precedented role in hydrocarbon metabolism clusters in Rhodococcus sp. and which may therefore participate in a β-myrcene degradation pathway in this organism.
Similar content being viewed by others

Introduction
The biotransformation of abundant, low-value renewable organic materials from plants to higher value compounds is an area of great contemporary interest, as the use of microbial or enzymatic catalysis can offer a route to natural-equivalent products which resonate with green technologies in industry (Chen et al. 2007; Woodley 2008). The C-10 monoterpenes represent one such source of potential renewable chemicals of value to the flavour and fragrance industry (Krings and Berger 1998; Vandamme and Soetaert 2002; Rozenbaum et al. 2006), with compounds such as limonene (de Carvalho and da Fonseca 2003; Duetz et al. 2003) carvone (de Carvalho and da Fonseca 2006) and menthol (Serra et al. 2005) having been studied extensively.
Hops, from the genus Humulus, are an abundant crop largely grown in central temperate regions and are widely cultivated for beer production. The lupulin glands from the flowers (cones) of the female Humulus lupulus plant are known to contain a complex mixture of C-10 terpenes as part of their natural essential oils, including the fragrant monoterpene alcohols linalool and geraniol (Kishimoto et al. 2005; Cheng et al. 2007). The acyclic monoterpene β-myrcene (7-methyl-3-methylene-1,6-octadiene; 1, Fig. 1) accounts for a relatively large fraction (up to 70%) of monoterpenes extracted from the essential oils of H. lupulus (from the Hop Union Directory of Hop Data, available at http://www.hopunion.com/education.shtml). β-Myrcene may therefore be considered as a relatively inexpensive starting material for biotransformations that target more commercially attractive derivatives.

Some biotransformations of β-myrcene in the literature. Pathway (a): to ipsdienol 2 in the bark beetle Ips pini (Sandstrom et al. 2006); (b): to three diol 3, 4 and 5 compounds by A. niger (Farooq et al. 2004; Yamazaki et al. 1988); (c): to α-(Z)-acaridiol 6 by Pleurotus ostreatus (Krings et al. 2008); (d): to myrcene 1,2-endoperoxide 7 and myrcene 1,4-endoperoxide 8 by Pleurotus spp. (Krugener et al. 2009); (e): to 2-methyl-6-methylen-2,7-octadien-1-ol 9 (myrcen-8-ol) by Pseudomonas sp. strain M1 (Iurescia et al. 1999); (f): to geraniol 10 in this study
β-Myrcene has been the subject of a number of biotransformation studies in the past (Ishida 2005), including descriptions of its metabolism in mammalian (de Oliveira et al. 1997; Madyastha 1987) and insect cells (Miyazawa and Murata 2000). One example of a natural biotransformation involving myrcene is found in the biosynthetic pathway of aggregation pheromones in the pine engraver beetle Ips pini, in which β-myrcene was demonstrated to be hydroxylated stereoselectively to (4R)-(−)-ipsdienol (2, Fig. 1; Sandstrom et al. 2006). β-myrcene has also been used as a substrate in microbial reactions. Biotransformation of 1 by Aspergillus niger yielded different 1, 2-diol compounds 3, 4 and 5 (Fig. 1) via the apparent epoxidation, followed by hydrolysis, of β-myrcene at each of the three double bonds (Farooq et al. 2004; Yamazaki et al. 1988). A wide range of fragrant monoterpene alcohols was also produced by incubation of 1 with submerged cultures of Ganoderma applanatum, Pleurotus flabellatus and Pleurotus sajor-caju (Busmann and Berger 1994). Further strains of Pleurotus have been shown to metabolise 1 to, in one instance, α-acaridiol 6 (Krings et al. 2008) and in another, two endoperoxide derivatives 7 and 8 (Krugener et al. 2009).
Reports of the biotransformation of β-myrcene by bacterial species are rare, but in one study, it was used as the sole source of carbon for growth by Pseudomonas sp. M1 (Iurescia et al. 1999). Resting cells of a β-myrcene negative mutant of M1, created using transposon mutagenesis and designated N22, accumulated 2-methyl-6-methylen-2,7-octadien-1-ol 9 (myrcen-8-ol) as the major metabolite of biotransformation of 1 (Fig. 1). The ability to grow on 1 was conferred on N22 through the transfer of a cosmid which contained four open-reading frames myrA, myrB, myrC and myrD, potentially encoding an aldehyde dehydrogenase, an alcohol dehydrogenase, an acyl-coenzyme A synthetase and an enoyl-CoA hydratase respectively. The identification of these genes enabled the authors to propose a pathway for degradation of 1 by Pseudomonas sp. M1.
Geraniol (10, Fig. 1) is a monoterpene alcohol with a wide range of uses in perfumery, food and also as an insect repellent. The global production of geraniol together with its derived esters was estimated recently to be 12,000 tonnes per year (Schwab et al. 2008). Whilst some of this material is extracted directly from natural products, the majority is based on an industrial chemical synthesis from naturally derived β-pinene. Selective abiotic routes from β-myrcene towards geraniol have also been described, through regioselective hydroxylation of Pd(II) (Takahashi et al. 1979) and cobalt (Howell and Pattenden 1990a, 1990b) complexes. Production of geraniol in any of these respects would preclude the potential use of the natural-equivalent label and so a direct biocatalytic route from β-myrcene to geraniol would be highly desirable. In this report, we describe the enrichment selection of a Rhodococcus strain designated MLT1 using β-myrcene as sole carbon source. Growth of the organism on β-myrcene induces the activity for biotransformation of the substrate to geraniol.
Materials and methods
Chemicals
Myrcene of 90% purity was purchased from Fluka. For use in enrichment and biotransformation experiments, the commercial compound was distilled by heating at 120°C under a reduced pressure of 15 mbar. Authentic geraniol (purity≥96%) and nerol (≥90%) standards were purchased from Fluka. A H. lupulus plant (Goldings variety) and soil surrounding the roots were obtained from Botanix Ltd.
Enrichment cultures and isolation of Rhodococcus erythropolis MLT1
Soil, root, leaf and flower samples from H. lupulus were homogenised individually in 20 mL of sterile water, 5 mL of which was used to inoculate 250-mL Erlenmeyer flasks containing 50 mL of M9 minimal medium (KH2PO4 3.1 g/L, K2HPO4 8.2 g/L, (NH4)2SO4 2.4 g/L, yeast extract 0.1 g/L, tryptone 0.1 g/L, MgSO4·7H2O 0.5 g/L, MnSO4·H2O 0.05 g/L, CaCl2·2H2O 0.01 g/L, molybdic acid 0.01 g/L, FeSO4·7H2O 2.5 g/L). The cultures were incubated aerobically at 25°C for 7 days with shaking at 150 rpm, before the addition of myrcene (1 g/L). After a further 12 days incubation, samples showing growth from the soil incubations were transferred (2% inoculum) to fresh M9 media (50 mL) with concentrations of myrcene at 1 and 5 g/L. At these concentrations, the myrcene was not completely soluble and tended to form emulsions. However, this did not seem to limit growth or result in uneven patterns of growth on the plates. After 6 days of growth, M9 agar plates containing myrcene (5 g/L) were inoculated with 100 μL dilutions (10−2–10−4) of this culture, and incubated at 30°C for 7 days. Plates were sub-cultured onto the same medium and incubated for a further 7 days, before picking individual colonies and plating again. Further incubation led to the appearance of a single strain. 16S rRNA sequence analysis of this strain was carried out by the National Collection of Industrial and Marine Bacteria (NCIMB, Aberdeen, Scotland). The strain has been deposited with NCIMB with the accession number NCIMB 14574.
Growth of R. erythropolis MLT1
To enable growth on β-myrcene in the vapour phase, adaptations of 250-mL Erlenmeyer flasks were made using glassblowing facilities. Glass tubing (50 mm in length, and 6 mm diameter) was attached to the internal base of the flask, the top of which protrudes 25 mm above the level of a 50 mL culture shaken at 150 rpm. Myrcene (0.5 mL) was added into this hollow central well allowing the vapour to fill into the headspace above the culture medium. For the monitoring of growth, 50-mL cultures of M9 media with either glucose 1.0 g/L, succinate 1.0 g/L, or myrcene (vapour phase) as sole carbon sources were inoculated from agar plates as described previously and incubated at 30°C for 24 h. The optical density (OD, measured at a wavelength of 600 nm) of these pre-cultures was measured prior to inoculation (10%) of three separate 50 mL cultures for each of glucose, succinate and myrcene. OD measurements were taken from each flask over a period of time, until stationary phase of the growth curve was reached.
Preparation of cell extracts
Cells were harvested using a Sorvall RC5B centrifuge equipped with an SS34 rotor (4°C, 20 min, 27,000×g) then resuspended in one-tenth growth volume of 50 mM potassium phosphate (KH2PO4 6.8 g/L, K2HPO4 8.7 g/L) buffer (pH 7.0). Cells were lysed by three passages through a continuous flow French press at 270 MPa. The cell debris was removed by centrifugation (4°C, 40 min, 34,500×g) before retaining the soluble cell extract, which was concentrated by ultracentrifugation using a Centricon with a 3,000-molecular-weight cut-off filter (Millipore, Amicon Ultra-15). The total concentration of protein was estimated after lysis using the method of Bradford (Bradford 1976).
Biotransformation of myrcene by R. erythropolis MLT1
R. erythropolis MLT1 was incubated in M9 media as described above, with myrcene as the sole carbon source administered in the vapour phase for 3 days. Harvested cells were washed twice with phosphate buffer, before resuspension (50 g/L) in 5 mL of buffer and the addition of myrcene (1 μL/mL, 7.4 mM). Controls with dead (autoclaved) cells, and myrcene (1 μL/mL) in buffer (no cells) were prepared under the same conditions. The samples were incubated for 1 h with shaking at 30°C. Cells were separated by centrifugation as before, and the solution saturated with NaCl before extracting three times with 5 mL of a 1:1 (petroleum ether:ethyl acetate) mixture. The organic layers were recombined, and dry MgSO4 added to remove excess water. The MgSO4 was filtered off, and the solution concentrated by leaving to evaporate overnight, before re-dissolving the resultant oil with 300 μL of a 1:1 petroleum ether:ethyl acetate mixture. For cytochrome P450 inhibition studies, cells were prepared using the methods previously mentioned, then pre-incubated with 1 and 5 mM concentrations of the inhibitors metyrapone and 1-aminobenzotriazole (ABT), shaking at 30°C for 0.5, 1 and 2 h before repeating the resting cell assay with myrcene.
Anaerobic resting cell assay
Resuspended cells were split into two aliquots. The standard resting cell assay with myrcene was repeated with one aliquot by incubating for 1 h at 25°C, whilst the other aliquot was stirred for 15 min in an anaerobic chamber to remove any dissolved oxygen from the solution prior to the addition of pre-equilibrated myrcene (1 μL/mL). The stirred solution was incubated for 1 h at 25°C under anaerobic conditions, and extracted with a pre-equilibrated 1:1 (petroleum ether:ethyl acetate) mixture. Samples were kept sealed airtight prior to GC analysis.
Metabolite identification
Gas chromatographic analysis of samples was performed routinely using an Agilent 6890 N gas chromatograph equipped with a J&W HP5 column (length 30 m, internal diameter 0.32 mm, film 0.25 μm). Two-microlitre samples were injected at 250°C in the split mode. For analysis of myrcene–geraniol biotransformations, an initial oven temperature of 60°C was increased through a 10°C/min gradient to 200°C, with a flow rate of the carrier gas helium at 10 mL/min. GC-MS analysis was performed with a Clarus 500 gas chromatograph (Perkin-Elmer) equipped with a J&W DB-1 column (30 m × 0.32 mm × 1 μm), coupled with a Clarus 500 mass spectrometer. One-microlitre samples were injected at 250°C in the split mode. An initial oven temperature of 60°C was held for 1.5 min before increasing at 4°C/min to 280°C with a further hold at this temperature for 15 min. The carrier gas helium was provided at a flow rate of 1.5 mL/min. The instrument was run in EI mode at an energy of 70 eV. Mass spectra were scanned in the range of 25 to 250 m/z.
Gel analysis of cell extracts and peptide MS analysis
Cell extract samples were diluted 1:1 in a loading buffer 50 mM Tris/HCl (pH 6.8), containing: glycerol 10%, SDS 2%, bromophenol blue 0.005% and β-mercaptoethanol 5%. Resolving gels (12% acrylamide) were set by adding tetramethylethylenediamine (TEMED) to a resolving gel buffer (1.5 M Tris, 0.4% SDS, pH 8.8) containing acrylamide and ammonium persulphate (APS). Stacking gels were poured from a 0.5 M Tris buffer (0.4% SDS, pH 6.8) containing acrylamide, APS and TEMED. A BioRad low molecular mass marker mixture from 14.4–97.4 kDa was used. Gels were stained with Coommasie Blue and destained using a 6:3:1 mixture of water: propan-2-ol: glacial acetic acid.
In-gel tryptic digestion was performed after reduction with dithioerythritol (DTE) and S-carbamidomethylation with iodoacetamide. Gel pieces were washed twice with 50% (v:v) aqueous acetonitrile containing 25 mM ammonium bicarbonate, and then once with acetonitrile before drying in a vacuum concentrator for 20 min. Sequencing-grade, modified porcine trypsin (Promega) was dissolved in 50 mM acetic acid, then diluted fivefold by adding 25 mM ammonium bicarbonate to give a final trypsin concentration of 0.01 μg/μL. Gel pieces were rehydrated by adding 10 μL of trypsin solution, and after 30 min, the gel pieces were submerged in a 25-mM ammonium bicarbonate solution. Digests were incubated overnight at 37°C. A 0.5-μL aliquot of each peptide mixture was applied directly to the ground steel MALDI target plate, followed immediately by an equal volume of a freshly prepared 5 g/L solution of 4-hydroxy-α-cyano-cinnamic acid (Sigma) in 50% aqueous (v:v) acetonitrile containing 0.1% , trifluoroacetic acid (v:v).
Positive-ion MALDI mass spectra were obtained using an Applied Biosystems 4700 Proteomics Analyzer (Applied Biosystems, Foster City, CA, USA) in reflectron mode, equipped with a Nd:YAG laser. MS spectra were acquired over a mass range of m/z 800–4,000. Final mass spectra were internally calibrated using the tryptic autoproteolysis products at m/z 842.509 and 2,211.104. Monoisotopic masses were obtained from centroids of raw, unsmoothed data. The 20 strongest peaks with a signal to noise greater than 40 were selected for CID-MS/MS analysis.
For CID-MS/MS, a Source 1 collision energy of 1 kV was used, with air as the collision gas. The precursor mass window was set to a relative resolution of 50, and the metastable suppressor was enabled. The default calibration was used for MS/MS spectra, which were baseline-subtracted (peak width 50) and smoothed (Savitsky–Golay with three points across a peak and polynomial order 4); peak detection used a minimum S/N of 5, local noise window of 50 m/z, and minimum peak width of 2.9 bins. Filters of S/N 20 and 30 were used for generating peak lists from MS and MS/MS spectra, respectively.
Tandem mass spectral data were submitted to database searching against the NCBI non-redundant database (20070926 version, 5519594 sequences, 1911975371 residues) and the Rhodococcus jostii RHA1 genome (http://www.rhodococcus.ca; 9161 sequences, 2893055 residues) using a locally running copy of the Mascot programme (Matrix Science Ltd., version 2.1), through the Applied Biosystems GPS Explorer software interface (version 3.6).
Where required MS/MS spectral data were also submitted to de novo sequencing using GPS Explorer TM software—DeNovo Explorer Version 3.6. The following parameters were used: Enzyme: trypsin, mass tolerance; 0.2 Da, fixed modifications; carbamidomethyl (C), variable modification; oxidation (M). The top ten sequence matches were submitted to an MSBlast search against the nrdb95 database using the facility provided by the European Molecular Biology Laboratory (Shevchenko et al. 2001).
Results
Enrichment selection and characterisation of R. erythropolis MLT1
R. erythropolis MLT1 was isolated from an enrichment culture using β-myrcene as a sole carbon source (SCS) inoculated initially with soil surrounding the roots of a hop plant, as described in the “Materials and methods” section, and identified through 16S rRNA sequence analysis (NCIMB). Growth of this strain in liquid media with myrcene (0.1–0.5 g/L) was very limited, and was likely to be a result of the poor solubility and toxicity observed at higher concentrations of myrcene. Improved growth was observed when the myrcene was supplied in the vapour phase in Erlenmeyer flasks that had been fitted with a central glass well, into which the β-myrcene was deposited. GC analysis of organic extracts of the growth medium at intervals showed an absence of β-myrcene, and it appears therefore that the substrate in the vapour phase is taken up from the flask headspace by the cells. In cultures using β-myrcene in the vapour phase as SCS a prolonged exponential phase of 15 h preceded a maximum OD (recorded at 600 nm) of 0.45 (Fig. 2). Growth rates were increased using glucose or succinate as SCS with exponential phases lasting 7 and 9 h, respectively, before reaching optical densities as measured by absorbance at 600 nm of 0.6 and 1.5, respectively (Fig. 2).

Growth curves plotting absorbance recorded at 600 nm (A 600) against time for R. erythropolis MLT1 utilising: succinate (filled diamond), glucose (filled square) and myrcene (filled upright triangle) as the sole sources of carbon for growth. Each point corresponds to the average value calculated on the basis of results recorded for three cultures grown in parallel
Biotransformation of β-myrcene
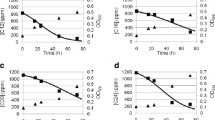
Resting cells of R. erythropolis MLT1 harvested from the late exponential phase of growth were resuspended (50 g/L) in a pH 7.0 phosphate buffer and incubated with myrcene (7.4 mM) for 1 h at 30°C (Fig. 3a). A variety of control experiments was performed that included incubating cells without myrcene, and incubating phosphate buffer and cells inactivated through autoclaving, with the substrate (Fig. 3e). No biotransformation products were observed in any of the controls; however, a single GC peak with retention time 7.60 min not present in the background was found to be produced only on incubation of β-myrcene-grown cells with β-myrcene. A time-course for this assay showed an increasing concentration of this product over an initial 5-min period (Fig. 3b–d), after which the concentration remained constant for approximately 2 h.

Chromatograms of injections of the myrcene biotransformation extracts after: a t = 1 h; b t = 0.1 min; c t = 0.5 min; d t = 5 min; e dead cell control at t = 1 h. β-myrcene eluted at 3.93 min and geraniol at 7.60 min
The dependence of this biotransformation on the presence of molecular oxygen was studied in a control experiment carried out under anaerobic conditions. A single batch of cells was prepared as before with myrcene as the SCS in the vapour phase. Division of the cell resuspension into two aliquots enabled reproduction of the standard biotransformation, again showing the formation of the peak at 7.60 min from 1 h incubation at a temperature of 25°C, replicating the ambient temperature measured in the anaerobic hood for the control assay. The second aliquot of active cells was pre-equilibrated in the anaerobic environment, and then challenged with the substrate. GC analysis of the anaerobic biotransformation mixture revealed no production of the peak at 7.60 min under anaerobic conditions.
Metabolite identification
The MS spectrum of the biotransformation product (Fig. 4) revealed good correlation to existing published data (Adams 1995; Jennings and Shibamoto 1980; Ojala et al. 1999) for the GC-MS analysis of geraniol (3,7-dimethylocta 2,6-dien-1-ol) 10 (Fig. 1). Evidence was also obtained to demonstrate that isomers of geraniol are not responsible for the major product peak. An authentic standard of the stereoisomer nerol ((2Z)-3,7-dimethylocta-2,6-dien-1-ol) run under the same GC conditions was found to have a HP5 retention time of 7.25–7.30 min compared to that for geraniol of 7.60 min. The structural isomer, isogeraniol ((3Z)-3,7-dimethylocta-3,6-dien-1-ol) has published retention indexes lower than those for nerol (Rocha et al. 2007). A calibration line generated from a geraniol standard shows an approximate 50 μM product concentration from the biotransformation in a 1-h assay representing a conversion from β-myrcene of ~2%. Further samples were prepared from three separate cultures for the biotransformation of myrcene each showing the product with a retention time of 7.60 min. In addition to these samples, three standards of geraniol at: 0.03, 0.05 and 0.5 mM were also prepared for further analytical work to confirm the product identity. A standard linear n-alkane series (C10–C18) was run on both DB5 (non-polar) and DB17 (polar) GC columns giving incremental retention times with increasing chain length. All samples and standards were then run on the DB17 column where a shift (increase) in retention time was observed for geraniol due to interactions of the hydroxyl group with the polar column, when compared to the standard alkane series (giving a retention time similar to the C14 alkane on the same column). The retention times of the three samples run on DB17 column (11.16–11.21 min) overlapped with those for the geraniol standards (11.13–11.20 min). Peak areas were proportional in samples and standards run on both HP5 and DB17 columns, again confirming the identity of the product peak as geraniol.

Mass spectrum for the major metabolite of biotransformation of β-myrcene by R. erythropolis MLT1, eluting at a retention time of 7.60 min in Fig. 3 and identified as geraniol
Cytochrome P450 inhibition studies
Cells were harvested from cultures grown in parallel and combined before splitting into separate aliquots to study the effects of cytochrome P450 inhibition on the biotransformation. GC analysis of the reactions catalysed by cells in the absence of inhibitors revealed concentrations of geraniol that were consistent with previous observations. Pre-incubation of cells at 30°C for 1 h with metyrapone led to a 23% reduction in the concentration of geraniol produced. The effect of incubation with 5 mM 1-aminobenzotriazole for the same period was a 73% decrease in the yield of geraniol. A range of concentrations from 0–5 mM was investigated for both inhibitors however there was no correlation observed between concentration of inhibitor and the negative effect on myrcene biotransformation. Extended pre-incubation times of up to 2 h with the inhibitors also did not result in greater levels of inhibition. Complete inhibition of the biotransformation of β-myrcene to geraniol was not observed for any of the assays conducted using cytochrome-P450 inhibitors.
Inducibility of β-myrcene biotransformation
The resting cell assay was repeated using cells grown on glucose or succinate as SCS. GC analysis of the extracts from these assays showed no geraniol product peak as previously described for those cells grown on β-myrcene as SCS. Insoluble and soluble extracts from cells grown on either glucose or β-myrcene were obtained and analysed by SDS-PAGE (Fig. 5a). Gel a clearly shows induced expression of a band of protein at a MW of approximately 16 kDa-labelled I. Analysis of tryptic digests of this band by mass spectrometry with subsequent comparison of peptide mass fragments against database searches using Mascot failed to identify any clear hits. Gel a also shows additional bands induced in the molecular mass region corresponding to between 43 and 66 kDa. Increased resolution of proteins in this region was achieved between these molecular masses (Fig. 5b) by decreasing the percentage acrylamide in the gel, and running for a longer period of time. At least three further bands, II, III and IV on the gel were enhanced in samples having been obtained from cells grown on β-myrcene. MS analysis of tryptic fragments obtained from bands II and IV followed by a BLAST search and comparison with entries in the Mascot database suggested II was homologous to the 60-kDa chaperonin GroEL from R. jostii RHA1 (Table 1). IV was assigned as a putative acyl-CoA dehydrogenase based on the similarity to a peptide fragment from Frankia alni (Table 1). Equivalent analysis of band III did not yield any clear hits within the Mascot database, however, de novo sequencing followed by searching against the BLAST database suggested that the protein was homologous to 2-hydroxymuconic acid semialdehyde dehydrogenase from Pseudomonas, based on the identification of peptide fragments LSYVEEAVSEGATLVTGG and DEFVAR.

SDS-PAGE analysis of the crude soluble cell extracts of R. erythropolis MLT1 grown on either glucose or β-myrcene as SCS. Gel a shows denatured proteins in the molecular mass range 14–97 kDa analysed on a 12% acrylamide gel: lane 1 BioRad low molecular mass markers with molecular mass shown in kDa; lane 2 Soluble extract from cells grown on glucose; lane 3, soluble extract from cells grown on β-myrcene; lane 4 Insoluble extract from cells grown on glucose; lane 5 Insoluble extract from cells grown on β-myrcene. Protein band labelled I could not be identified using mass spectrometric analysis of tryptic fragments. Gel b shows denatured proteins in the molecular mass range 45–97 kDa analysed on an 8% acrylamide gel: lane 6 BioRad low molecular mass markers with molecular mass shown in kDa; lane 7 soluble extract from cells grown on glucose; lane 8, soluble extract from cells grown on β-myrcene. MS analysis of tryptic fragments suggested that proteins II, III and IV may be homologues of a GroEL-like chaperone, an aldehyde dehydrogenase and an acyl-CoA dehydrogenase, respectively
Discussion
Little is known about the enzymatic systems that exist for the metabolism of β-myrcene in bacteria. In this report, a strain of R. erythropolis was isolated that is observed in resting cell biotransformations to convert β-myrcene to the important flavour compound geraniol. The chemical-enzymatic mechanism of this transformation is at this stage unclear, and an explanation must await labelling experiments to reveal if the oxygen atom introduced originates from molecular oxygen, and is thus catalysed by an oxygenase; or water, and thus catalysed by a lyase, each discussed below.
Much is known about the mechanisms by which some bacteria oxygenate alkene substrates (Ensign 2001), but these mechanisms routinely involve epoxidation by a multicomponent monooxygenase enzyme, of which different types have been observed in, for example Xanthobacter strain Py2 (Small and Ensign 1997) and Rhodococcus strain AD45 (Van Hylckama Vlieg et al. 2000). No component of such a monooxygenase was yet identified in 1-D gels of extracts resulting from cells induced with β-myrcene from mass spectrometric analysis of tryptic fragments, although the biotransformation of β-myrcene by Rhodococcus MLT1 clearly required an aerobic environment. Another possible route for the incorporation of a single atom of molecular oxygen into an alkene substrate was described in styrene-oxidising bacteria, in which the successive action of styrene monooxygenase, followed by an isomerase enzyme, results in the terminally monooxygenated compound phenylacetaldehyde from styrene oxide (Panke et al. 1998), but as yet, no such system has been described in the metabolism of aliphatic alkenes. Of other enzymes that catalyse the incorporation of a single atom of oxygen into an alkene substrate, the contribution of cytochrome-P450 in the oxygenation of β-myrcene by MLT1 has not been absolutely ruled out by the relevant inhibition studies. Such alkenes are certainly substrates for cyt-P450 (Sandstrom et al. 2006), although in previous studies, such as those on the cytochrome-P450 steroid 9-α hydroxylase from Mycobacterium fortuitum (Kang and Lee 1997), the level of inhibition by metyrapone increased with increasing inhibitor concentration. This was not the case for the transformation by MLT1 presented herein.
The chemical transformation of β-myrcene to geraniol is formally a hydration reaction. The hydration of myrcene could also lead to linalool, and, whilst there is a precedent for the microbial transformation of linalool to geraniol through a putative 3,1-hydroxyl-∆1-∆2-mutase (Foß and Harder 1997), few appropriate lyase enzymes have been described, to our knowledge, that might hydrate the double bond of an alkene that was not conjugated, as part of an α, β-unsaturated system conjugated to a ketone or (thio)ester. One example was the limonene hydratase described by Oriel and co-workers, which was used to convert limonene to α-terpineol and carvone (Savithiry et al. 1997). The other chemical methods of conversion described in the “Introduction” section are dependent on, for example, hydrocobaltation of the terminal alkene, followed by radical trapping with TEMPO and subsequent reduction with zinc (Howell and Pattenden 1990a). Whilst these mechanisms of hydroxylation have no formal equivalent in biochemistry, the use of metal-assisted catalysis for the transformation of β-myrcene by MLT1 cannot at this stage be ruled out.
Of the enzymes that are apparently induced in MLT1 by growth on β-myrcene, each has precedent in inducible systems for hydrocarbon degradation in bacteria described previously. An aldehyde dehydrogenase of theoretical molecular mass 54.3 kDa, similar to that observed in Fig. 5, gel b, was one of the proteins encoded in a putative operon for β-myrcene degradation in Pseudomonas sp. M1 (Iurescia et al. 1999). A 13-fold upregulation of expression of the GroEL chaperone proteins was observed in Rhodococcus sp. RHA1 when induced by propane, the degradation of which is also dependent on a multicomponent monooxygenase (Sharp et al. 2007). Given the established pathways for the degradation of geraniol by, for example, Pseudomonas, through the formation and subsequent degradation of a geranyl-CoA thioester (Förster-Fromme et al. 2006), the involvement of an acyl-CoA dehydrogenase as suggested by MS analysis of induced proteins, would also not be unexpected; indeed an acyl-CoA dehydrogenase with specificity for citronellyl-CoA was recently described (Förster-Fromme et al. 2008). The unambiguous identification of the enzymes responsible for degradation of β-myrcene in MLT1 awaits the cloning of the relevant genes and characterisation of their expressed products.
Enzymes involved in the metabolism of β-myrcene by bacteria may, in the future, be usefully applied as biocatalysts in the production of natural-equivalent flavour and fragrance compounds. Control experiments for the biotransformation described in this report have demonstrated that geraniol is a biogenic product of β-myrcene incubation with R. erythropolis MLT1 rather than an artefact of effects due to reaction medium, inactive biological material or pH. The one-pot biotransformation of β-myrcene to geraniol might present therefore a potentially attractive industrial biocatalytic route towards a fragrant monoterpene, from an inexpensive and naturally abundant hydrocarbon starting material. We are currently exploring the nature of the key enzymes involved in β-myrcene metabolism by R. erythropolis MLT1, with a view both to proposing a mechanism for the biotransformation, and to developing a scaleable process for geraniol production.
References
Adams RP (1995) Identification of essential oil components by gas chromatography – mass spectrometry. Allured, IL
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Busmann D, Berger RG (1994) Conversion of myrcene by submerged cultured basidiomycetes. J Biotechnol 37:39–43
Chen ZM, Liu JH, Tao JH (2007) Biocatalysis for green chemistry and drug development. Prog Chem 19:1919–1927
Cheng AX, Lou YG, Mao YB, Lu S, Wang LJ, Chen XY (2007) Plant terpenoids: biosynthesis and ecological functions. J Integr Plant Biol 49:179–186
de Carvalho C, da Fonseca MMR (2003) Towards the bio-production of trans-carveol and carvone from limonene: induction after cell growth on limonene and toluene. Tetrahedron Asymmetr 14:3925–3931
de Carvalho C, da Fonseca MMR (2006) Carvone: why and how should one bother to produce this terpene. Food Chem 95:413–422
de Oliveira A, Ribiero-Pinto L, Otto S (1997) Induction of liver monooxygenases by beta-myrcene. Toxicology 124:135–140
Duetz WA, Bouwmeester H, van Beilen JB, Witholt B (2003) Biotransformation of limonene by bacteria, fungi, yeasts, and plants. Appl Microbiol Biotechnol 61:269–277
Ensign SA (2001) Microbial metabolism of aliphatic alkenes. Biochemistry 40:5845–5853
Farooq AR, Rahman A, Choudhary AI (2004) Fungal transformation of monoterpenes. Curr Org Chem 8:353–367
Förster-Fromme K, Chattopadhyay A, Jendrossek D (2008) Biochemical characterization of AtuD from Pseudomonas aeuruginosa, the first member of a new subgroup of acyl-CoA dehydrogenases for citronellyl-CoA. Microbiology 154:789–796
Förster-Fromme K, Höschle B, Mack C, Bott M, Armbruster W, Jendrossek D (2006) Identification of genes and proteins necessary for catabolism of acyclic terpenes and leucine/isovalerate in Pseudomonas aeuruginosa. Appl Environ Microbiol 72:4819–4828
Foβ S, Harder J (1997) Microbial transformation of a tertiary allylalcohol: regioselective isomerisation of linalool to geraniol without nerol formation. FEMS Microbiol Lett 149:71–75
Howell AR, Pattenden G (1990a) Hydrocobaltation reactions of 1, 3-dienes - regioselective hydroxylation of myrcene to geraniol and to (+/-)-linalool via allylcobaloxime intermediates. J Chem Soc, Perkin Trans 1:2715–2719
Howell AR, Pattenden G (1990b) Regioselective hydroxylations of 1, 3-dienes via hydrocobaltation reactions—facile conversion of myrcene to geraniol and to (+/−)-linalool. J Chem Soc Chem Commun 2:103–104
Ishida T (2005) Biotransformation of terpenoids by mammals, microorganisms, and plant-cultured cells. Chem Biodivers 2:569–590
Iurescia S, Marconi AM, Tofani D, Gambacorta A, Paterno A, Devirgiliis C, van der Werf MJ, Zennaro E (1999) Identification and sequencing of beta-myrcene catabolism genes from Pseudomonas sp strain M1. Appl Environ Microbiol 65:2871–2876
Jennings W, Shibamoto T (1980) Qualitative analysis of flavour and fragrance volatiles by glass capillary gas chromatography. Academic, New York
Kang HK, Lee SS (1997) Heterogeneous natures of the microbial steroid 9α-hydroxylase in nocardioforms. Arch Pharm Res 20:519–524
Kishimoto T, Wanikawa A, Kagami N, Kawatsura K (2005) Analysis of hop-derived terpenoids in beer and evaluation of their behavior using the stir bar-sorptive extraction method with GC-MS. J Agric Food Chem 53:4701–4707
Krings U, Berger RG (1998) Biotechnological production of flavours and fragrances. Appl Microbiol Biotechnol 49:1–8
Krings U, Hapetta D, Berger RG (2008) A labeling study on the formation of perillene by submerged cultured oyster mushroom, Pleurotus ostreatus. Appl Microbiol Biotechnol 78:533–541
Krugener S, Schaper C, Krings U, Berger RG (2009) Pleurotus species convert monoterpenes to furanoterpenoids through 1, 4-endoperoxides. Biores Technol 100:2855–2860
Madyastha KM (1987) Metabolism of beta-myrcene in-vivo and in-vitro - its effects on rat-liver microsomal-enzymes. Xenobiotica 17:539–549
Miyazawa M, Murata T (2000) Biotransformation of beta-myrcene by the larvae of common cutworm (Spodoptera litura). J Agric Food Chem 48:123–125
Ojala M, Ketola RA, Mansikka T, Kotiaho T, Kostiainen R (1999) Determination of mono- and sesquiterpenes in water samples by membrane inlet mass spectrometry and static headspace gas chromatography. Talanta 49:179–188
Panke S, Witholt B, Schmid A, Wubbolts MG (1998) Towards a biocatalyst for (S)-styrene oxide production: characterization of the styrene degradation pathway of Pseudomonas sp. strain VLB120. Appl Environ Microbiol 64:2032–2043
Rocha SM, Coelho E, Zrostlikova J, Delgadillo I, Coimbra MA (2007) Comprehensive two-dimensional gas chromatography with time-of-flight mass spectrometry of monoterpenoids as a powerful tool for grape origin traceability. J Chromatogr A 1161:292–299
Rozenbaum HF, Patitucci ML, Antunes OAC, Pereira N (2006) Production of aromas and fragrances through microbial oxidation of monoterpenes. Br J Chem Eng 23:273–279
Sandstrom P, Welch WH, Blomquist GJ, Tittiger C (2006) Functional expression of a bark beetle cytochrome P450 that hydroxylates myrcene to ipsdienol. Insect Biochem Mol Biol 36:835–845
Savithiry N, Cheong TK, Oriel P (1997) Production of alpha-terpineol from Escherichia coli cells expressing thermostable limonene hydratase. Appl Biochem Biotechnol 63–65:213–220
Schwab W, Davidovich-Rikanati R, Lewinsohn E (2008) Biosynthesis of plant-derived flavor compounds. Plant J 54:712–732
Serra S, Fuganti C, Brenna E (2005) Biocatalytic preparation of natural flavours and fragrances. Trends Biotechnol 23:193–198
Sharp JO, Sales CM, LeBlanc JC, Liu J, Wood TK, Eltis LD, Mohn WW, Alvarez-Cohen L (2007) An inducible propane monooxygenase is responsible for N-nitrosodimethylamine degradation by Rhodococcus sp strain RHA1. Appl Environ Microbiol 73:6930–6938
Shevchenko A, Sunyaev S, Loboda A, Shevchenko A, Bork P, Enz W, Standing KG (2001) Charting the proteomes of organisms with unsequenced genomes by MALDI quadrupole time-of-flight mass spectrometry and BLAST homology searching. Anal Chem 73:1917–1926
Small FJ, Ensign SA (1997) Alkene monooxygenase from Xanthobacter strain Py2. J Biol Chem 272:24913–24920
Takahashi M, Suzuki H, Morooka Y, Ikawa T (1979) Regioselective hydroxylation of beta-myrcene using Pd(II) complexes. Chem Lett 8:53–56
Van Hylckama Vlieg JET, Leemhuis H, Lutje Spelberg JH, Janssen DB (2000) Characterisation of the gene cluster involved in isoprene metabolism in Rhodococcus sp. strain AD45. J Bacteriol 182:1956–1963
Vandamme EJ, Soetaert W (2002) Bioflavours and fragrances via fermentation and biocatalysis. J Chem Technol Biotechnol 77:1323–1332
Woodley JM (2008) New opportunities for biocatalysis: making pharmaceutical processes greener. Trends Biotechnol 26:321–327
Yamazaki Y, Hayashi Y, Hori N, Mikami Y (1988) Microbial conversion of β-myrcene by Aspergillus niger. Agric Biol Chem 52:2921–2922
Acknowledgments
We are grateful to Botanix Ltd, Paddock Wood, Kent, U.K. and to the Biotechnology and Biological Sciences Research Council for funding a CASE studentship to M.L.T. We are also grateful to Botanix for providing plant material, chemicals and GC-MS services.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Thompson, M.L., Marriott, R., Dowle, A. et al. Biotransformation of β-myrcene to geraniol by a strain of Rhodococcus erythropolis isolated by selective enrichment from hop plants. Appl Microbiol Biotechnol 85, 721–730 (2010). https://doi.org/10.1007/s00253-009-2182-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-009-2182-6