
Abstract
The amount of available light plays a key role in the growth and development of microbial communities. In the present study, we tested to what extent sponge-associated prokaryotic communities differed between specimens of the sponge species Cinachyrella kuekenthali and Xestospongia muta collected in dimly lit (caves and at greater depths) versus illuminated (shallow water) habitats. In addition to this, we also collected samples of water, sediment, and another species of Cinachyrella, C. alloclada. Overall, the biotope (sponge host species, sediment, and seawater) proved the major driver of variation in prokaryotic community composition. The light habitat, however, also proved a predictor of compositional variation in prokaryotic communities of both C. kuekenthali and X. muta. We used an exploratory technique based on machine learning to identify features (classes, orders, and OTUs), which distinguished X. muta specimens sampled in dimly lit versus illuminated habitat. We found that the classes Alphaproteobacteria and Rhodothermia and orders Puniceispirillales, Rhodospirillales, Rhodobacterales, and Thalassobaculales were associated with specimens from illuminated, i.e., shallow water habitat, while the classes Dehalococcoidia, Spirochaetia, Entotheonellia, Nitrospiria, Schekmanbacteria, and Poribacteria, and orders Sneathiellales and Actinomarinales were associated with specimens sampled from dimly lit habitat. There was, however, considerable variation within the different light habitats highlighting the importance of other factors in structuring sponge-associated bacterial communities.
Similar content being viewed by others


Introduction
The widespread application of DNA sequencing technologies has revealed a diversity of microorganisms far greater than previously thought. This has led to a fundamental shift in our understanding of animal-microbe associations with multicellular organisms no longer being considered fully autonomous entities but holobionts [1]. Sponges, one of the oldest multicellular animal lineages, have been extensively studied as model holobionts in marine environments. In addition to being model organisms of the early evolution of host-microbe symbioses, sponges also play important structural roles in coral reefs where they provide living environments for several other marine species and bridge the benthic and pelagic zones with their filtering activities [2]. They are also prolific sources of bioactive natural products, many of which are produced by their microbial symbionts [3, 4].
Several spatial and environmental processes, e.g., temperature, salinity, chlorophyll a concentrations, and the degree of light illumination, affect the composition and functioning of microbial communities including those associated with sponges [5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20]. Light is of particular importance to marine microbial communities. In the marine environment, light levels decrease from brightly illuminated surface waters to more dimly lit conditions at greater depths and, in caves, from the cave entrance to the dark recesses of the cave interior. This reduction in light levels has a direct impact on several environmental processes in aquatic environments [21,22,23,24,25].
Here, we compared prokaryotic communities of X. muta and one species of Cinachyrella (C. kuekenthali) sampled in dimly lit (caves and at greater depths) and illuminated (shallow water) habitats surrounding the island of Martinique. Both of these species span a gradient from shallow to mesophotic waters thereby providing models to study the effects of light attenuation on sponge-associated prokaryotic communities [13, 26, 27]. In addition to this, we also sampled sediment, water, and specimens of another shallow water Cinachyrella species, C. alloclada. The giant barrel sponge X. muta has been previously assigned high microbial abundance (HMA) status [28,29,30]. It is one of the most abundant species in Caribbean reefs [31]. Sponges of the genus Cinachyrella are common in the coastal waters of Florida and the Caribbean. Cinachyrella alloclada and Cinachyrella kuekenthali are sympatric species in Martinique and also elsewhere in the wider Caribbean basin [32]. However, unlike the shallow water species C. alloclada, C. kuekenthali has been observed along a depth range from 0.2 to 100 m [33]. Species of the genus Cinachyrella are often covered by sediment and algae and can look very similar; they can, thus, be challenging to differentiate in the field without careful skeletal observation. Members of this genus appear to have marked differences in their associated microbial communities [34].
The main underlying hypothesis of the present study is that sponges in different light habitats host distinct prokaryotic communities. In addition to this, we aimed to assess if different sponge species, sediment, and seawater also housed distinct prokaryotic communities, and to identify taxa associated with different light habitats in the sponge species X. muta.
Materials and Methods
Sample Collection
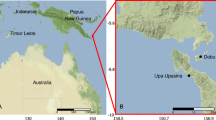
In the present study, we collected 34 samples of three sponge species, four sediment, and three water samples between 3 and 90 m depth from various sites around the island of Martinique (Fig. 1) from September 5th till October 10th, 2016. The shallow water and cave samples were collected between 3 and 31 m depth using SCUBA diving or snorkeling and the samples from deeper water were collected by dredging. The deeper samples consisted of three C. kuekenthali and nine X. muta specimens. These were all sampled from depths greater than 60 m. The dredge consisted of a triangular metal frame of 30 cm with a 1 mm mesh net, which was connected to a rope with a length of 200 m and pulled by a light boat. Each dredge lasted between 5 and 20 min depending on the type of background. Sponges were handpicked from the dredge, photographed and preserved in 96% ethanol. Additional data on all samples collected including the geographical coordinates of the sample sites and depths are provided in Supplementary data 1.

Map of Martinque showing the locations of the sampling sites
Twelve samples of Xestospongia muta (Schmidt, 1870) were collected in shallow waters between 7 and 31 m, nine samples were collected in deeper waters, > 60 m, and three were collected from shallow caves between 14 and 21 m depth. In the present study, we pooled the cave specimens and specimens sampled at greater depths in order to distinguish dimly lit versus illuminated (shallow water) habitats. Specimens from caves and deeper water are, however, distinguished in the figures. Xestospongia muta is found along a shallow to mesophotic depth gradient [13]. Olson and Gao [14] initially identified X. muta as hosting a diverse microbial community including abundant Chloroflexi and cyanobacterial components. Four individuals of Cinachyrella alloclada (Uliczka, 1929) were collected between 3 and 22 m depth. Three individuals of Cinachyrella kuekenthali (Uliczka, 1929) were collected in shallow waters between 3 and 22 m and three in deeper waters (> 60 m). Photographs of selected sponge specimens are shown in Fig. 2.

Selected photographs of the focal sponge species., A Cinachyrella alloclada, B Cinachyrella kuekenthali sampled from shallow water, C deep water, Xestospongia muta sampled from D deep water, E a cave, and F shallow water. The samples in C and D were collected using dredging and the pictures were taken in the remote lab
Four sediment samples were collected from the top 5 cm surface layer in sites between 10 and 15 m depth using a Falcon tube. Three seawater samples were collected from sites between 2 and 5 m depth; 1 l was filtered through a Millepore white Isopore membrane filter (0.22 μm pore size) to obtain seawater prokaryotic communities. All samples were stored in ethanol (96%) and kept in a cooler until they were finally stored at −20 °C at the Naturalis Biodiversity Center. Voucher specimens of each sponge sample have been deposited at the sponge collection of the Naturalis Biodiversity Center, Leiden, The Netherlands (as RMNH POR).
DNA Extraction and Next-Generation Sequencing Analysis
PCR-ready genomic DNA was isolated from all samples using the FastDNA® SPIN soil Kit (MPbiomedicals) following the manufacturer’s instructions. Briefly, the whole membrane filters, for water samples, were cut into small pieces and transferred to Lysing Matrix E tubes containing a mixture of ceramic and silica particles; for the other samples, ± 500 mg of sponge tissue (including parts of the surface and interior) or sediment was added. The microbial cell lysis was performed in the FastPrep® Instrument (Q Biogene) for 80 s at 6.0 ms−1. The extracted DNA was eluted into DNase/Pyrogen-Free water to a final volume of 50 μl and stored at −20 °C until use. The 16S rRNA gene V3-V4 variable region PCR primers 341F 5’-CCTACGGGNGGCWGCAG-3’ and 785R 5’-GACTACHVGGGTATCTAATCC-3’ [35] with barcode on the forward primer were used in a 30 cycle PCR assay using the HotStarTaq Plus Master Mix Kit (Qiagen, USA) under the following conditions: 94 °C for 3 min, followed by 28 cycles of 94 °C for 30 s, 53 °C for 40 s, and 72 °C for 1 min, after which a final elongation step at 72 °C for 5 min was performed. After amplification, PCR products were checked on a 2% agarose gel to determine the success of amplification and the relative intensity of bands; the blank control did not yield any bands. PCR product was used to prepare the DNA library following the Illumina TruSeq DNA library preparation protocol. Next-generation, paired-end sequencing was performed at MrDNA (Molecular Research LP; http://www.mrdnalab.com/; last checked 18 November 2016) on an Illumina MiSeq device (Illumina Inc., San Diego, CA, USA) following the manufacturer’s guidelines. Sequences from each end were joined following Q25 quality trimming of the ends followed by reorienting any 3′–5′ reads back into 5′–3′ and removal of short reads (< 150 bp).
16S rRNA Gene Sequencing Analysis
The 16S rRNA gene amplicon libraries were analyzed using QIIME2 (version 2019.7) [36]. Raw data was imported yielding a demultiplexed “qza” data file (artifact). The DADA2 plugin [37] in QIIME 2 was subsequently used to trim sequences (final length 400 nt). The DADA2 analysis yielded output archives containing an OTU (also known as amplicon sequence variant or “ASV”) table, denoising stats and a fasta file of representative sequences. The feature-classifier plugin with the extract-read method was then used with the i-sequences argument set to silva-138-99-seqs.qza. This was followed by the feature-classifier plugin with the fit-classifier-naive-bayes method and the i-reference-taxonomy method set to silva-138-99-tax.qza. Both silva-138 files can be obtained from https://docs.qiime2.org/2020.8/data-resources/?highlight=silva. The feature-classifier plugin was then used with the classify-sklearn method and the i-reads argument set to the representative sequences file generated by the DADA2 analysis to produce a table with taxonomic assignment for all OTUs. Finally, mitochondria, chloroplasts, and Eukaryota were filtered out using the qiime taxa plugin with the filter-table method. The OTU and taxonomy tables were later merged in R [38]. The DNA sequences generated in this study can be downloaded from NCBI BioProject Id: PRJNA715755.
Statistical Analyses
A table containing the OTU counts was imported into R using the read.csv function. Supplementary data 2 contains all operational taxonomic unit (OTU) counts per sample and taxonomic assignments of all OTUs. The 50 most abundant OTUs are presented in Supplementary data 3. The raw OTU counts matrix was used to compare diversity among groups and to test for differences in higher taxon abundances. Diversity indices were obtained using the rarefy and diversity functions from the vegan [39] package in R. Evenness was calculated by dividing Shannon’s H′ by the number of OTUs in each sample. We tested for significant differences in the relative abundance of the most abundant prokaryotic phyla and classes, OTU richness, evenness, Shannon’s H′, and Fisher’s alpha indices among groups (sediment, seawater, C. alloclada, and C. kuekenthali and X. muta from dimly lit and illuminated habitats thus forming seven groups/biotopes) with an analysis of deviance using the glm function in R. For the diversity indices, we set the family argument to “tweedie” using the tweedie function [40] in R with var.power=1.5 and link.power=0 (a compound Poisson–gamma distribution). For the relative abundances of higher taxa, we set the family argument to quasibinomial. Using the GLM model, we tested for significant variation among groups using the ANOVA function in R with the F test. We used the emmeans function in the emmeans library [41] in R to perform multiple comparisons of mean abundance among groups using estimated marginal means (EMM) with the false discovery rate (fdr) method in the adjust argument of the emmeans function and a P value of 0.05. Only groups with at least three samples were included.
Variation in prokaryotic composition among groups was assessed with principal coordinates analysis (PCO). We tested for significant differences among groups with an adonis analysis from the vegan package. For the PCO, a Bray-Curtis distance matrix was first obtained using the Phyloseq package [41] and the count data was rarefied using the rarefy_even_depth function with the sample.size argument set to the minimum sample size (13038 in the present study) and subsequently log10 transformed. A separate analysis was run including only specimens of X. muta in order to test for a significant effect of light habitat on prokaryotic composition.
We used an exploratory technique based on machine learning, Boruta, to identify features, which distinguished between X. muta specimens sampled in dimly lit (deeper and cave) versus illuminated (shallow water) habitat. Boruta, named after a slavic forest demon, is a random forest wrapper, which is used to evaluate feature importance [42]. The first step in the Boruta algorithm is the generation of a data frame consisting of randomly shuffled versions of all original features, which are termed shadow features. This data frame is then attached to the data frame of original features creating a new data frame with double the amount of features. A random forest machine learning algorithm is then applied to the new data frame consisting of all the original and shadow features in addition to the response variable. The importance of each original feature is then compared with a threshold based on the shadow features. This process is repeated a given number of times (permutations) producing a binomial distribution. Original features at the tails of the distribution are either rejected or accepted while those in the middle are considered tentative. Increasing the number of randomizations can help to resolve tentatively classified features. In the present study, we generated three data frames consisting of classes, orders, OTUs, and a response variable indicating whether a sample of X. muta was collected in dimly lit versus illuminated habitat. Prior to the Boruta analysis, highly correlated features (> 0.8) were removed from the data frames. The Boruta function from the Boruta package in R [43] was then used with the habitat (dimly lit versus illuminated) as response variable and orders, classes, or OTUs as features (predictive variables). The doTrace argument in the Boruta function was set to 2 and the maxRuns argument set to 1000; other arguments used default values. The number of significant features was identified and the six features (classes, orders, or OTUs) with the highest importance presented.
Results
After quality control and removal of OTUs assigned to chloroplasts and mitochondria, the data set consisted of 3172098 sequences and 11977 OTUs. In terms of sequences, the most abundant phyla in the present study were Proteobacteria (1371433 sequences, 4556 OTUs), Chloroflexi (442626 sequences, 602 OTUs), Actinobacteriota (351143 sequences, 572 OTUs), and Myxococcota (143938 sequences, 314 OTUs). Rarefied richness and evenness were significantly higher in sediment than all other biotopes (Fig. 3 and Supplementary data 4). Evenness was also significantly higher in X. muta than Cinachyrella spp. Richness, in turn, was significantly lower in X. muta sampled from both dimly lit and illuminated habitat than in both Cinachyrella species. Shannon’s H′ and Fisher’s alpha diversity indices largely reflected results of evenness and rarefied richness, respectively (Supplementary data 4).

Boxplots showing values for selected diversity indices. Results of GLM analyses are shown after the respective index. Evenness: F6,34 = 56.48, P < 0.001, richness: F6,34 = 46.61, P < 0.001, Shannon: F6,34 = 35.78, P < 0.001, and Fisher: F6,34 = 65.65, P < 0.00. The x-axis labels refer to sediment (Sd), water (Wt), Cinachyrella alloclada (Ca), Cinachyrella kuekenthali from dimly lit (Cd) and illuminated (Cl) habitats, and Xestospongia muta from dimly lit (Xd) and illuminated (Xl) habitats. Colored symbols indicate specimens collected from caves (Cv), deep (Dp), and shallow water (Sh)
The eight most abundant phyla, in terms of sequences, together accounted for 86.3% of all sequences and 60.57% of all OTUs (Fig. 4). Boxplots showing variation in the relative abundances of the six most abundant phyla and classes are presented in Figs. 5 and 6, respectively. The relative abundance of Proteobacteria was significantly lower in X. muta than in both Cinachyrella species (Supplementary data 5). In contrast, the relative abundances of Chloroflexi and Myxococcota were significantly higher in C. kuekenthali and X. muta, sampled from dimly lit and illuminated habitat, than in all other groups/biotopes. There were no significant differences in the relative abundance of Actinobacteriota among groups. The relative abundances of Acidobacteriota and Gemmatimonadota were significantly higher in sediment, C. kuekenthali and X. muta than in C. alloclada or seawater (Supplementary data 5).

Stacked barplots of the mean relative abundances of the most abundant phyla. The x-axis labels refer to sediment (Sd), water (Wt), Cinachyrella alloclada (Ca), Cinachyrella kuekenthali from dimly lit (Cd) and illuminated (Cl) habitat, and Xestospongia muta from dimly lit (Xd) and illuminated (Xl) habitat

Boxplots showing the relative abundances for the most abundant phyla. Results of GLM analyses are shown after the respective phylum. Proteobacteria: F6,34 = 22.11, P < 0.001, Chloroflexi: F6,34 = 51.05, P < 0.001, Actinobacteriota: F6,34 = 1.42, P = 0.235, Myxococcota: F6,34 = 28.34, P < 0.001, Acidobacteriota: F6,34 = 12.83, P < 0.001, and Gemmatimonadota: F6,34 = 40.41, P < 0.001. The x-axis labels refer to sediment (Sd), water (Wt), Cinachyrella alloclada (Ca), Cinachyrella kuekenthali from dimly lit (Cd) and illuminated (Cl) habitats, and Xestospongia muta from dimly lit (Xd) and illuminated (Xl) habitats. Colored symbols indicate specimens collected from caves (Cv), deep (Dp), and shallow water (Sh)

Boxplots showing the relative abundances for the most abundant classes. Results of GLM analyses are shown after the respective class. Gammaproteobacteria: F6,34 = 25.39, P < 0.001, Alphaproteobacteria: F6,34 = 30.3, P < 0.001, Acidimicrobiia: F6,34 = 1.5, P = 0.207, Dehalococcoidia: F6,34 = 37.53, P < 0.001, bacteriap25: F6,34 = 41.24, P < 0.001, and Anaerolineae: F6,34 = 15.91, P < 0.001. The x-axis labels refer to sediment (Sd), water (Wt), Cinachyrella alloclada (Ca), Cinachyrella kuekenthali from dimly lit (Cd) and illuminated (Cl) habitats, and Xestospongia muta from dimly lit (Xd) and illuminated (Xl) habitats. Colored symbols indicate specimens collected from caves (Cv), deep (Dp), and shallow water (Sh)
At the class level, the relative abundance of Gammaproteobacteria was significantly higher in C. kuekenthali than all other groups and the relative abundance of Alphaproteobacteria was significantly higher in C. alloclada and seawater than all other groups (Supplementary data 6). The relative abundances of Dehalococcoidia and bacteriap25 were significantly higher in C. kuekenthali and X. muta from dimly lit and illuminated habitat than in all other groups. There was no significant difference in the relative abundance of Acidimicrobiia between pairs of groups (Supplementary data 6).
There was clear clustering of samples based on biotope (sediment, seawater, and sponge species). The first axis of the ordination separated X. muta from the remaining biotopes. The second axis, in turn, separated C. kuekenthali from seawater samples with sediment samples and specimens of C. alloclada intermediate (Fig. 7). Specimens of C. kuekenthali from dimly lit and illuminated habitats also separated along axis 2. The third axis of variation separated sediment from remaining samples while the fourth axis of variation separated C. alloclada from C. kuekenthali and seawater samples (Supplementary data 7). The group/biotope proved a significant predictor of variation in composition (adonis: F6,34 = 11.78, R2 = 0.675, P < 0.001). When only including the X. muta specimens (Fig. 8), the light habitat (dimly lit versus illuminated) also proved a significant predictor of variation in composition (adonis: F1,22 = 2.28, R2 = 0.094, P < 0.001).

Ordination showing the first two axes of the principal coordinates analysis (PCO) of OTU composition. Symbols are color coded and represent samples from different groups as shown in the legend on the right side of the figure. Gray symbols represent weighted averages scores for OTUs. The symbol sizes for OTUs are proportional to their abundances (number of sequence reads). The eigenvalues for the first and second axes were 5.54 and 0.90, respectively, and explained 48.3 and 7.8%, respectively, of the total variation in the data. The symbols refer to sediment (Sd), water (Wt), Cinachyrella alloclada (Ca), Cinachyrella kuekenthali from dimly lit (Cd) and illuminated (Cl) habitats, and Xestospongia muta from dimly lit (Xd) and illuminated (Xl) habitats

Ordination showing the first two axes of the principal coordinates analysis (PCO) of OTU composition including only samples of X. muta collected from caves, deeper, and shallow water. Symbols are color coded and represent samples from different groups as shown in the legend on the right side of the figure. Gray symbols represent weighted averages scores for OTUs. The symbol sizes for OTUs are proportional to their abundances (number of sequence reads). The eigenvalues for the first and second axes were 0.21 and 0.16, respectively, and explained 12.4 and 9.4%, respectively, of the total variation in the data. The symbols refer to Xestospongia muta specimens sampled from caves (Cv), deep (Dp), and shallow water (Sh)
Using only X. muta specimens, the Boruta analysis yielded nine significant predictive classes and eleven significant predictive orders. Six of the classes and orders with the greatest importance values are presented in Figs. 9 and 10. Other significant predictors included the classes Alphaproteobacteria and Dehalococcoidia (Fig. 6). The classes Alphaproteobacteria and Rhodothermia had greater median relative abundances in shallow water X. muta specimens and the remaining classes in specimens sampled from dimly lit habitat. At the order level, all of the alphaproteobacterial orders, with the notable exception of the Sneathiellales, had greater median relative abundances in shallow water specimens. Median Actinomarinales relative abundance was greater in specimens sampled from dimly lit habitat.

Boxplots showing the relative abundances of bacterial classes, which differentiated between deeper water and cave versus shallow water sponges. The x-axis labels refer to Xestospongia muta specimens sampled from dimly lit (Xd) and illuminated (Xl) habitats. Colored symbols indicate specimens collected from cave (Cv), deep (Dp), and shallow water (Sh)

Boxplots showing the relative abundances of bacterial orders, which differentiated between deeper water and cave versus shallow water sponges. The x-axis labels refer to X. muta specimens sampled from dimly lit (Xd) and illuminated (Xl) habitats. Colored symbols indicate specimens collected from caves (Cv), deep (Dp), and shallow water (Sh)
There was considerable variation in relative abundance among groups of the 50 most abundant OTUs (Fig. 11). The prokaryotic community of C. alloclada differed from seawater and other groups in the higher relative abundances of eight OTUs assigned to the Actinobacteriota (OTU-135), NB1-j (OTUs 50, 94, and 211), Alpha- (OTUs 4, 78) and Gammaproteobacteria (OTUs 136 and 180). Cinachyrella kuekenthali housed greater abundances of OTUs assigned to the Actinobacteriota (OTU-29), Chloroflexi (OTU-141), Entotheonellaeota (OTU-149), Myxococcota (OTU-127), Nitrospirota (OTU-98), Spirochaetota (OTU-86), and Gammaproteobacteria (OTUs 6, 32, 38, and 44). OTUs 6, 32, and 44 were all assigned to the AqS1 genus (Nitrosococcales). The OTUs of X. muta were more evenly distributed between dimly lit and illuminated (shallow water) habitats and differed most with respect to the other groups by the abundance of a set of OTUs assigned to the Chloroflexi and three OTUs assigned to the Myxococcota (26, 117, and 191). These OTUs were absent or recorded in very low abundances in the other groups/biotopes. OTUs 106, 132, 197, 208, and 221 were all assigned to the Caldilineales order (Anaerolineae), whereas OTUs 67, 97, and 146 were assigned to the SAR202 clade (Dehalococcoidia). Twenty-two OTUs proved significant predictors of specimens of X. muta sampled in dimly lit versus illuminated habitat. OTUs 173, 252, and 746 assigned to the Rhodobacterial genera Albidovulum, and Silicimonas and the gammaproteobacterial family Woeseiaceae, respectively, were more abundant in shallow water specimens (Fig. 12). In contrast, the OTUs 640, 1376, and 1755, assigned to the Actinomarinales order, AqS1 genus (Nitrosococcales), and SAR202 clade, respectively, were more abundant in specimens from dimly lit habitat (Fig. 12). Only three OTUs assigned to the Cyanobacteria were included in the 50 most abundant OTUs. OTUs 3 and 20 were assigned to the genus Synechococcus and Prochlorococcus, respectively. Both of these were most abundant in seawater and C. alloclada and were also more abundant in shallow water specimens of C. kuekenthali and X. muta. OTU-154 was assigned to Synechococcus spongiarum and reached greatest abundance in shallow water samples of X. muta, where it accounted for 1.22% of the total community compared to 0.14% in deep water specimens and 0.38% in cave specimens. It was not recorded in C. kuekenthali.

Relative abundance of the most abundant OTUs color coded according to prokaryotic phylum. The x-axis labels refer to sediment (Sd), water (Wt), Cinachyrella alloclada (Ca), Cinachyrella kuekenthali from dimly lit (Cd) and illuminated (Cl) habitats, and Xestospongia muta from dimly lit (Xd) and illuminated (Xl) habitat. The circle size of the OTU is proportional to the mean percentage of sequences per biotope as indicated by the symbol legend at the right of the figure below the phylum assignment legend

Boxplots showing the relative abundances of OTUs, which differentiated between deeper water and cave versus shallow water sponges. The x-axis labels refer to: Xestospongia muta specimens sampled from dimly lit (Xd) and illuminated (Xl) habitats. Colored symbols indicate specimens collected from caves (Cv), deep (Dp) and shallow water (Sh)
Discussion
In the present study, we showed that the prokaryotic communities of three Caribbean sponge species were compositionally distinct from one another and from sediment and seawater. This result is in line with a large number of previous studies highlighting the importance of host identity in structuring prokaryotic community composition [44,45,46,47,48]. The sponges C. kuekenthali and X. muta housed greater relative abundances of Chloroflexi, Myxococcota, and lower abundances of Alphaproteobacteria than sediment, seawater, and the sponge C. alloclada. Xestospongia muta was, furthermore, particularly enriched with Chloroflexi members in both the Dehalococcoidia and Anaerolineae classes while Gammaproteobacteria were particularly abundant in C. kuekenthali.
In addition to the above, we also showed that specimens of both C. kuekenthali and X. muta housed compositionally distinct prokaryotic communities in different light habitats. Chloroflexi, for example, were more abundant in specimens of X. muta sampled from dimly lit than illuminated habitats, while the reverse held for Alphaproteobacteria. The PCO analysis including only X. muta specimens showed that specimens sampled from caves and deeper water largely overlapped, but both were distinct from specimens sampled from shallow water habitat. This suggests that the importance of depth in structuring prokaryotic communities may primarily be related to light attenuation, as opposed to other factors, which vary with depth, such as temperature, at least over the depth range sampled in the present study.
Elsewhere in the Caribbean (Lee Stocking Island, Bahamas, and Little Cayman, Cayman Islands), Morrow et al. [13] studied populations of X. muta in shallow water and mesophotic reefs. Using stable isotopes, Morrow et al. [13] showed a change in stable isotope enrichment with depth indicative of a shift from photoautotrophy to heterotrophy. They, furthermore, identified reductions in the relative abundances of Alphaproteobacteria (Rhodobacteraceae and Rhodospirillaceae), Synechococcaceae, and Rhodothermaceae with depth and increased abundances of Entotheonellaceae, Chloroflexi, Poribacteria, and Ectothiorhodospiraceae (Gammaproteobacteria; Chromatiales), although this differed between sites. At one site, SAR202 and Anaerolineae members were abundant members of the microbiome across the whole depth range with the exception of the most shallow samples. At the other site, in contrast, OTUs assigned to the genus Synechococcus were abundant with the exception of the deepest specimens where Chloroflexi members dominated thereby highlighting the potential importance of local factors in depth-related shifts in composition across sites. Synechococcus members have generally high nitrogen requirements. Morrow et al. [13] attributed the relative stability in Cyanobacteria with depth to increasing NOx concentrations at the site in question such that the increased nitrogen availability compensated for reduced irradiance. They suggested that Chloroflexi may be responding to different environmental parameters at the different sites including photoautotrophy and competition for inorganic carbon with Synechococcus members at one site and a shift to other, still-to-be-identified, nutrient sources at the other site, thus explaining the different responses to depth between sites.
Although Morrow et al. [13] observed shifts in composition with depth, the core members of the bacterial communities of X. muta remained relatively stable between 10 to 91 m depth. These core members were assigned to the phyla Chloroflexi, Cyanobacteria (Synechococcus), Proteobacteria, Actinobacteria, Acidobacteria, Poribacteria, and Thaumarchaeota (Nitrosopumilus), and are believed to play key roles in their sponge hosts with respect to host defense and fulfilling nutrient requirements [13]. Synechococcus members (including S. spongiarum), for example, have been shown to transfer carbon to their host sponges [49].
The results of our study largely confirm those of Morrow et al. [13] in terms of the reduced relative abundance of Alphaproteobacteria with depth and enrichment with other groups such as the Dehalococcoidia, Entotheonellaeota, and Poribacteria suggesting a shift from phototrophy to other functions, e.g., use of dissolved organic matter. In contrast to Morrow et al. [13], the genus Synechococcus was only a minor component of the prokaryotic community of X. muta in our study, but was more abundant in illuminated than dimly lit habitat.
In order to delve deeper into our data and identify prokaryotic taxa (classes, orders, and OTUs) associated with dimly lit versus illuminated habitat in the sponge X. muta, we used an exploratory technique (Boruta) based on the random forest algorithm. The Boruta algorithm can play an important role in studies using next-generation sequencing given the large amount of data involved and a tendency to focus on the most abundant components of the microbial communities. In the present study, use of the Boruta algorithm enabled us to identify a number of predictors at differing levels of taxonomic resolution, some of which may otherwise have been ignored. It is important to remember, however, that the present study is only observational in nature and future studies will be required to explore the mechanisms involved in the host-prokaryotic associations presented here.
The median relative abundances of the classes Alphaproteobacteria and Rhodothermia and orders Puniceispirillales, Rhodospirillales, Rhodobacterales, and Thalassobaculales were greater in specimens from illuminated than dimly lit habitat; the reverse held for the classes Dehalococcoidia, Spirochaetia, Entotheonellia, Nitrospiria, Schekmanbacteria, Poribacteria, and orders Sneathiellales and Actinomarinales.
Rhodothermia, assigned to the Bacteroidota, were mainly represented by the class Rhodothermales and family Rhodothermaceae in the present study. Rhodothermaceae members have been previously obtained from seawater and were reported to carry rhodopsin genes [50]. Another strain obtained from seawater was reported to be gram-negative, obligately aerobic, heterotrophic, and catalase-positive and capable of nitrate reduction [51].
The orders Rhodobacterales, Puniceispirillales, and Rhodospirillales are all members of the Alphaproteobacteria class, a widespread, dominant, and diverse group of free-living and host-associated bacteria [48, 52,53,54]. The order Rhodobacterales includes members capable of photosynthesis under anoxic conditions [55, 56] and they have been observed in high abundances in corals [45, 57, 58] and sponges [53, 59]. In the present study, Rhodospirillales members were more abundant in seawater than in X. muta, indicating that it is possible that they may be part of the transient prokaryotic community of the sponge, rather than the symbiotic host microbiome. Rhodospirillales are commonly found in bacterioplankton communities of coral reefs [60]. They are phototrophic microorganisms [61], which fits with their greater abundance in shallow water specimens. In the present study, the most abundant Puniceispirillales OTUs were either assigned to the SAR116 clade or to the genus Constrictibacter (Supplementary Data 3). Members of the SAR116 clade have been previously reported from oligotrophic seawater [62], whereas members of Constrictibacter were more abundant in sponges [62]. The prevalence of the SAR116 clade in the bacterioplankton community of seawater [63] suggests that its presence in the sponge microbiome may also be transient. Organisms belonging to SAR116 are normally more abundant in the euphotic zone of the ocean and are known to possess the photoprotein proteorhodopsin [64]. Members of the genus Constrictibacter have been reported to transmit vertically in multiple species of the tropical sponge genus Ircinia [65] in addition to other eukaryotes [66]. The functional role of the genus Constrictibacter is still poorly understood due to the lack of known strains [67].
Dehalococcoidia (phylum Chloroflexi) members have been recorded across varying biotopes including terrestrial soils [68], marine subsurface sediments [69], and sponges [70,71,72]. Dehalococcoidia members comprise obligate organohalide-respiring species, which is an anaerobic bacterial respiratory process that uses halogenated hydrocarbons as terminal electron acceptors during electron transport [73]. Halogenated hydrocarbons have been detected in various marine sponges, e.g., Aplysina aerophoba, Agelas wiedenmayeri, and Agelas conifera, where they have been suggested to play roles in chemical defense mechanisms including antibiotics and signaling molecules [70, 72]. In the present study, most OTUs assigned to the Dehalococcoidia were also assigned to the SAR202 clade at order level. Busch et al. [74] identified two core OTU members of deep sea sponges using a 90% sequence similarity threshold of which one belonged to the Chloroflexi-Dehalococcoidia-SAR202 clade-hydrothermal vent metagenome and the other to the Actinobacteriota-Acidimicrobiia-Microtrichales-Microtrichaceae-Sva0996 marine group.
The class Schekmanbacteria (assigned to the phylum Schekmanbacteria) was first detected in the metagenome of sediments in the proximity of freshwater aquifers [75]. However, since then, members have been detected in several metagenomes from various marine environments, including in association with marine sponges [6, 76, 77]. Interestingly, multiple studies have reported an increasing presence of Ca. Schekmanbacteria organisms with depth [6, 76]. Functionally, Schekmanbacteria members are thought to be involved in sulfur cycling, as a number of studies have detected genes related to sulfate oxidation and reduction in the genomes of these organisms [78,79,80].
Members of the class Spirochaetia are frequently reported in association with tropical marine invertebrates, including corals [45, 46, 81] and sponges [45, 46, 82,83,84,85]. In sponges of the genera Tsitsikamma and Cyclacanthia, a bacterium assigned to the genus Spirochaeta (class Spirochaetia) was a key member of their core microbial communities and was suggested to have co-evolved with its respective hosts [86]. Others have likewise found a strong cophylogenetic signal between members of the class Spirochaetia and various coral and sponge hosts [81], which suggests that they are symbionts as opposed to transient organisms. Although implied in the production of secondary metabolites [86], little is known regarding the ecological role of Spirochaetia in sponge hosts. In non-marine hosts such as termites, members of the genus Treponema (class Spirochaetia) were suggested to function as nitrogen fixers [87], while in the marine environment, Spirochaeta appears to metabolize carbon sources in the gills of lucinid clams [88].
In the present study, levels of Entotheonellaeota were similar for X. muta and C. kuekenthali, but the phylum was practically absent from sediment, water, and C. alloclada. In X. muta, the median relative abundance was greater in dimly lit than illuminated habitat. Recently, Ruiz et al. [89] found that the bacteria Candidatus Entotheonella, a member of this phylum, was among the dominant morphotypes observed in the mesohyl and larvae of the cave dwelling sponge Plakina kanaky [89]. The authors suggested that members of this order might be vertically transmitted in P. kanaky and, due to their ability to produce unique bioactive compounds, confer an adaptive advantage to the sponge [89]. In fact, there is strong evidence that the genus is responsible for the production of biologically active compounds in the sponge Theonella swinhoei, which is known to be a prolific producer of unique bioactive natural products [90]. Together with the work of Ruiz et al. [89], our results hint that members of the Entotheonellaeota phylum may have a relevant ecological role for sponges inhabiting dimly lit habitat.
Poribacteria members were initially associated with a set of sponge species, mainly belonging to the order Verongida [91]. More recently, however, members of the phylum have been found in several other sponge species and other hosts such as corals [92,93,94,95,96,97]. Among sponges, Poribacteria have mainly been associated with HMA sponges [29, 98]. In the present study, poribacterial abundance was higher in samples of C. kuekenthali sampled at greater depths, and was very low in sediment, seawater, and C. alloclada (Supplementary data 8).
Members of the alphaproteobacterial order Sneathiellales deviated from the main trend in the class by having a greater median relative abundance in sponge specimens from dimly-lit habitat. Sneathiellales members have been previously detected in tidal mudflats [99], marine sediment and water [100,101,–102], as well as in association with nudibranch and coral hosts [103, 104]. The nudibranch-associated Sneathiellales were halotolerant, aerobic, and chemoheterotrophic [103]. Sneathiellales members of Mediterranean seawater communities were shown to be involved in the degradation of polycyclic aromatic hydrocarbons [105]. It is unknown what role they may play in their sponge hosts.
Previous studies have observed greater abundances of selected taxa in shallow, euphotic waters. These included Flavobacterales, Rhodobacterales, Actinomarinales, Verrucomicrobiales, Cellvibrionales, and SAR86. In contrast, SAR11, SAR324, SAR202, UBA10353 marine group, Marinimicrobia, Thiomicrospirales, Nitrospinales, and Nitrosopumilaceae were observed to increase in relative abundance with depth [15, 102, 106]. Our results with respect to depth-related differences in members of X. muta, appear to, at least partly, overlap observations of depth-related differences in bacterioplankton communities. Note that in the present study, water samples were only sampled from shallow water habitat. In future studies, it would be interesting to sample water from dimly lit habitat in order to assess if potentially transient sponge microbiome members shift in line with the local bacterioplankton community.
Conclusion
Here, our results showed that the sponge species C. kuekenthali and X. muta hosted compositionally distinct prokaryotic communities in different light environments. With respect to X. muta, our results are in line with a previous study on the impact of depth on bacterial communities of X. muta in the Bahamas [13]. Interestingly, our dataset showed that the median relative abundance of the order Entotheonellales was greatest in Xestospongia muta specimens collected from dimly lit habitat. This observation and the recent study by Ruiz et al. [89] suggest that members of the Entotheonellales order may play an important ecological role for sponges inhabiting these environments. Members of this order are known for their ability to produce unique bioactive compounds with potential for drug discovery. Future work should clarify this apparent pattern and test if caves, for example, are a hotspot of sponge-associated Entotheonellales with biotechnological potential.
Data Availability
Information about the availability of the data reported in this work has been included in the “Material and Methods” section.
References
McFall-Ngai M, Hadfield MG, Bosch TC, Carey HV, Domazet-Lošo T, Douglas AE, Dubilier N, Eberl G, Fukami T, Gilbert SF, Hentschel U, King N, Kjelleberg S, Knoll AH, Kremer N, Mazmanian SK, Metcalf JL, Nealson K, Pierce NE et al (2013) Animals in a bacterial world, a new imperative for the life sciences. Proc Natl Acad Sci USA 110:3229–3236. https://doi.org/10.1073/pnas.1218525110
Li Z, Hentschel U, Webste N, Olson J, Häggblom M (2020) Editorial: Special issue on sponge microbiome. FEMS Microbiol Ecol 96:fiaa075. https://doi.org/10.1093/femsec/fiaa075
Indraningrat AA, Smidt H, Sipkema D (2016) Bioprospecting sponge-associated microbes for antimicrobial compounds. Mar Drugs 2:87. https://doi.org/10.3390/md14050087
Mehbub MF, Lei J, Franco C, Zhang W (2014) Marine sponge derived natural products between 2001 and 2010: trends and opportunities for discovery of bioactives. Mar Drugs 12:4539–4577. https://doi.org/10.3390/md12084539
Astudillo-García C, Bell JJ, Montoya JM, Moitinho-Silva L, Thomas T, Webster NS, Taylor MW (2020) Assessing the strength and sensitivity of the core microbiota approach on a highly diverse sponge reef. Environ Microbiol 22:3985–3999. https://doi.org/10.1111/1462-2920.15185
Busch K, Hanz U, Mienis F, Mueller B, Franke A, Roberts EM, Rapp HT, Hentschel U (2020) On giant shoulders: how a seamount affects the microbial community composition of seawater and sponges. Biogeosciences 17:3471–3486. https://doi.org/10.5194/bg-17-3471-2020
Chow CE, Sachdeva R, Cram JA, Steele JA, Needham DM, Patel A, Parada AE, Fuhrman JA (2013) Temporal variability and coherence of euphotic zone bacterial communities over a decade in the Southern California Bight. ISME J 7:2259–2273. https://doi.org/10.1038/ismej.2013.122
Cleary DFR, Polónia ARM, Swierts T, Coelho FJRC, de Voogd NJ, Gomes NCM (2022) Spatial and environmental variables structure sponge symbiont communities. Mol Ecol 31:4932–4948. https://doi.org/10.1111/mec.16631
Dobretsov SV, Gosselin LA, Qian PY (2010) Effects of solar PAR and UV radiation on tropical biofouling communities. Mar Ecol Prog Ser 402:31–43. https://doi.org/10.3354/meps0845
Gómez-Consarnau L, González JM, Coll-Lladó M, Gourdon P, Pascher T, Neutze R, Pedrós-Alió C, Pinhassi J (2007) Light stimulates growth of proteorhodopsin-containing marine Flavobacteria. Nature 11:210–213. https://doi.org/10.1038/nature05381
Huq A, West PA, Small EB, Huq MI, Colwell RR (1984) Influence of water temperature, salinity, and pH on survival and growth of toxigenic Vibrio cholerae serovar 01 associated with live copepods in laboratory microcosms. Appl Environ Microbiol 48:420–424. https://doi.org/10.1128/aem.48.2.420-424.1984
Johnson CN, Bowers JC, Griffitt KJ, Molina V, Clostio RW, Pei S, Laws E, Paranjpye RN, Strom MS, Chen A, Hasan NA, Huq A, Noriea 3rd NF, Grimes DJ, Colwell RR (2012) Ecology of Vibrio parahaemolyticus and Vibrio vulnificus in the coastal and estuarine waters of Louisiana, Maryland, Mississippi, and Washington (United States). Appl Environ Microbiol 78:7249–7257. https://doi.org/10.1128/AEM.01296-12
Morrow KM, Fiore CL, Lesser MP (2016) Environmental drivers of microbial community shifts in the giant barrel sponge, Xestospongia muta, over a shallow to mesophotic depth gradient. Environ Microbiol 18:2025–2038. https://doi.org/10.1111/1462-2920.13226
Olson JB, Gao X (2013) Characterizing the bacterial associates of three Caribbean sponges along a gradient from shallow to mesophotic depths. FEMS Microbiol Ecol 85:74–84. https://doi.org/10.1111/1574-6941
Rodríguez-Ramos T, Nieto-Cid M, Auladell A, Guerrero-Feijóo E, Varela MM (2021) Vertical niche partitioning of Archaea and Bacteria linked to shifts in dissolved organic matter quality and hydrography in North Atlantic Waters. Front Mar Sci 8:673171. https://doi.org/10.3389/fmars.2021.673171
Saw JHW, Nunoura T, Hirai M, Takaki Y, Parsons R, Michelsen M, Longnecker K, Kujawinski EB, Stepanauskas R, Landry Z, Carlson CA, Giovannoni SJ (2020) Pangenomics analysis reveals diversification of enzyme families and niche specialization in globally abundant SAR202 bacteria. mbio 11:e02975–e02919. https://doi.org/10.1128/mBio.02975-19
Stauder M, Vezzulli L, Pezzati E, Repetto B, Pruzzo C (2010) Temperature affects Vibrio cholerae O1 El Tor persistence in the aquatic environment via an enhanced expression of GbpA and MSHA adhesins. Environ Microbiol Rep 2:140–144. https://doi.org/10.1111/j.1758-2229.2009.00121.x
Steffen K, Indraningrat AAG, Erngren I, Haglöf J, Becking LE, Smidt H, Yashayaev I, Kenchington E, Pettersson C, Cárdenas P, Sipkema D (2022) Oceanographic setting influences the prokaryotic community and metabolome in deep-sea sponges. Sci Rep 12:3356. https://doi.org/10.1038/s41598-022-07292-3
Steinert G, Taylor MW, Deines P, Simister RL, de Voogd NJ, Hoggard M, Schupp PJ (2016) In four shallow and mesophotic tropical reef sponges from Guam the microbial community largely depends on host identity. PeerJ 4:e1936. https://doi.org/10.7717/peerj.1936
Steinert G, Busch K, Bayer K, Kodami S, Arbizu PM, Kelly M, Mills S, Erpenbeck D, Dohrmann M, Wörheide G, Hentschel U, Schupp PJ (2020) Compositional and quantitative insights into bacterial and archaeal communities of South Pacific deep-sea sponges (Demospongiae and Hexactinellida). Front Microbiol 11:716. https://doi.org/10.3389/fmicb.2020.00716
Hader DP (2000) Effects of solar UV-B radiation on aquatic ecosystems. Adv Space Res 26:2029–2040. https://doi.org/10.1016/s0273-1177(00)00170-8
Church MJ, Ducklow HW, Karl DM (2004) Light dependence of [3H]leucine incorporation in the oligotrophic North Pacific ocean. Appl Environ Microbiol 70:4079–4087. https://doi.org/10.1128/AEM.70.7.4079-4087.2004
Klaus JS, Janse I, Heikoop JM, Sanford RA, Fouke BW (2007) Coral microbial communities, zooxanthellae and mucus along gradients of seawater depth and coastal pollution. Environ Microbiol 9:1291–1305. https://doi.org/10.1111/j.1462-2920.2007.01249.x
Karlsson J, Byström P, Ask J, Ask P, Persson L, Jansson M (2009) Light limitation of nutrient-poor lake ecosystems. Nature 23:506–509. https://doi.org/10.1038/nature08179
Hunt DE, Lin Y, Church MJ, Karl DM, Tringe SG, Izzo LK, Johnson ZI (2013) Relationship between abundance and specific activity of bacterioplankton in open ocean surface waters. Appl Environ Microbiol 79:177–184. https://doi.org/10.1128/AEM.02155-12
Olson JB, Kellogg CA (2010) Microbial ecology of corals, sponges, and algae in mesophotic coral environments. FEMS Microbiol Ecol 73:17–30. https://doi.org/10.1111/j.1574-6941.2010.00862.x
Carella M, Agell G, Cárdenas P, Uriz MJ (2016) Phylogenetic reassessment of Antarctic Tetillidae (Demospongiae, Tetractinellida) reveals new genera and genetic similarity among morphologically distinct species. PLoS One 24:e0160718. https://doi.org/10.1371/journal.pone.0160718
Gloeckner V, Wehrl M, Moitinho-Silva L, Gernert C, Schupp P, Pawlik JR, Lindquist NL, Erpenbeck D, Wörheide G, Hentschel U (2014) The HMA-LMA dichotomy revisited: an electron microscopical survey of 56 sponge species. Biol Bull 227:78–88. https://doi.org/10.1086/BBLv227n1p78
Moitinho-Silva L, Steinert G, Nielsen S, Hardoim CCP, Wu Y-C, McCormack GP, López-Legentil S, Marchant R, Webster N, Thomas T, Hentschel U (2017) Predicting the HMA-LMA Status in Marine Sponges by Machine Learning. Front Microbiol 8. https://doi.org/10.3389/fmicb.2017.00752
Montalvo NF, Hill RT (2011) Sponge-associated bacteria are strictly maintained in two closely related but geographically distant sponge hosts. Appl Environ Microbiol 77:7207–7216. https://doi.org/10.1128/AEM.05285-11
Bayona LM, Kim MS, Swierts T, Hwang GS, de Voogd NJ, Choi YH (2021) Metabolic variation in Caribbean giant barrel sponges: influence of age and sea-depth. Mar Environ Res 172:105503. https://doi.org/10.1016/j.marenvres.2021.105503
Cárdenas P, Menegola C, Rapp HT, Díaz MC (2009) Morphological description and DNA barcodes of shallow-water Tetractinellida (Porifera: Demospongiae) from Bocas del Toro, Panama, with description of a new species. Zootaxa 2276:1–39. https://doi.org/10.11646/zootaxa.2276.1.1
Fernandez JCC, Rodriguez PRD, Santos GG, Pinheiro U, Muricy G (2018) Taxonomy of deep-water tetillid sponges (Porifera, Demospongiae, Spirophorina) from Brazil, with description of three new species and new characters. Zootaxa 4429:53–88. https://doi.org/10.11646/ZOOTAXA.4429.1.2
Cuvelier ML, Blake E, Mulheron R, McCarthy PJ, Blackwelder P, Thurber RL, Lopez JV (2014) Two distinct microbial communities revealed in the sponge Cinachyrella. Front Microbiol 4:581. https://doi.org/10.3389/fmicb.2014.00581
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO (2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucl Acids Res 41:e1–e1. https://doi.org/10.1093/nar/gks808
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Caporaso JG (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol 37(8):852–857. https://doi.org/10.1038/s41587-019-0209-9
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. https://doi.org/10.1038/nmeth.3869
R Core Team (2022). R: a language and environment for statistical Computing. https://www.R-project.org/
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H (2020) Vegan: community ecology package. https://CRAN.R-project.org/package=vegan.
Tweedie MC (1984, December) An index which distinguishes between some important exponential families. In Statistics: Applications and new directions: Proc. Indian statistical institute golden Jubilee International conference 579:579–604
McMurdie PJ, Holmes S (2013) phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 8(4):e61217. https://doi.org/10.1371/journal.pone.0061217
Kursa MB, Jankowski A, Rudnicki W (2010) Boruta - a system for feature selection. Fundam Inform 101:271–285. https://doi.org/10.3233/FI-2010-288
Kursa MB, Rudnicki WR (2010) Feature selection with the Boruta package. J Stat Softw 36:1–13. https://doi.org/10.18637/jss.v036.i11
Chaib De Mares M, Jiménez DJ, Palladino G, Gutleben J, Lebrun LA, Muller EEL, Wilmes P, Sipkema D, van Elsas JD (2018) Expressed protein profile of a Tectomicrobium and other microbial symbionts in the marine sponge Aplysina aerophoba as evidenced by metaproteomics. Sci Rep 8:11795. https://doi.org/10.1038/s41598-018-30134-0
Cleary DFR, Polónia ARM, Reijnen BT, Berumen ML, de Voogd NJ (2020) Prokaryote communities inhabiting endemic and newly discovered sponges and octocorals from the Red Sea. Microb Ecol 80:103–119. https://doi.org/10.1007/s00248-019-01465-w
Cleary DFR, Polónia ARM, de Voogd NJ (2021) Composition and diversity of prokaryotic communities sampled from sponges and soft corals in Maldivian waters. Mar Ecol 42:e12638. https://doi.org/10.1111/maec.12638
Ribes M, Dziallas C, Coma R, Riemann L (2015) Microbial diversity and putative diazotrophy in high- and low-microbial-abundance mediterranean sponges. Appl Environ Microbiol 81:5683–5693. https://doi.org/10.1128/AEM.01320-15
Thomas T, Moitinho-Silva L, Lurgi M, Björk JR, Easson C, Astudillo-García C, Olson JB, Erwin PM, López-Legentil S, Luter H, Chaves-Fonnegra A, Costa R, Schupp PJ, Steindler L, Erpenbeck D, Gilbert J, Knight R, Ackermann G, Victor Lopez J et al (2016) Diversity, structure and convergent evolution of the global sponge microbiome. Nat Commun 7:11870. https://doi.org/10.1038/ncomms11870
Erwin PM, Thacker RW (2008) Cryptic diversity of the symbiotic cyanobacterium Synechococcus spongiarum among sponge hosts. Mol Ecol 17:2937–2947. https://doi.org/10.1111/j.1365-294x2008.03808.x
Nakajima Y, Yoshizawa S, Park S, Kumagai Y, Wong SK, Ogura Y, Hayashi T, Kogure K (2017) Draft genome sequence of Rubricoccus marinus SG-29T, a marine bacterium within the family Rhodothermaceae, which contains two different rhodopsin genes. Genome Announc 5:e00990–e00917. https://doi.org/10.1128/genomeA.00990-17
Park S, Yoshizawa S, Kogure K, Yokota A (2011) Rubricoccus marinus gen. nov., sp. nov., of the family ‘Rhodothermaceae’, isolated from seawater. Int J Syst Evol Microbiol 61:2069–2072. https://doi.org/10.1099/ijs.0.026294-0
Cleary DFR, Polónia ARM, Huang YM, Putchakarn S, Gomes NCM, de Voogd NJ (2019) A comparison of prokaryote communities inhabiting sponges, bacterial mats, sediment and seawater in Southeast Asian coral reefs. FEMS Microbiol Ecol 1:fiz169. https://doi.org/10.1093/femsec/fiz169
Karimi E, Keller-Costa T, Slaby BM, Cox CJ, da Rocha UN, Hentschel U, Costa R (2019) Genomic blueprints of sponge-prokaryote symbiosis are shared by low abundant and cultivatable Alphaproteobacteria. Sci Rep 13:1999. https://doi.org/10.1038/s41598-019-38737-x
Williams KP, Sobral BW, Dickerman AW (2007) A robust species tree for the alphaproteobacteria. J Bacteriol 189:4578–4586. https://doi.org/10.1128/JB.00269-07
Luo H, Moran MA (2014) Evolutionary ecology of the marine Roseobacter clade. Microbiol Mol Biol Rev 78:573–587. https://doi.org/10.1128/MMBR.00020-14
Kaftan D, Medova H, Selyanin V, Kopejtka K, Koblizek M (2019) Extended temperature optimum of photosynthetic reaction centers in Rhodobacterales. Photosynthetica 57:361–366. https://doi.org/10.32615/ps.2019.039
Appah JKM, Dillane E, Lim A, O’Riordan R, O’Reilly L, Macedo L, Wheeler AJ (2021) Cold-water coral microbiome and environmental microbial communities in a remote NE Atlantic submarine canyon setting: microbial diversity, coral health and prospects. Preprint (Version 1) available at Research Square. https://doi.org/10.21203/rs.3.rs-192160/v1
Sunagawa S, DeSantis TZ, Piceno YM, Brodie EL, DeSalvo MK, Voolstra CR, Weil E, Andersen GL, Medina M (2009) Bacterial diversity and White Plague Disease-associated community changes in the Caribbean coral Montastraea faveolata. ISME J 3:512–521. https://doi.org/10.1038/ismej.2008.131
Cleary DFR, Polónia ARM (2020) Marine lake populations of jellyfish, mussels and sponges host compositionally distinct prokaryotic communities. Hydrobiologia 847:3409–3425. https://doi.org/10.1007/s10750-020-04346-3
Frade PR, Glasl B, Matthews SA, Mellin C, Serrão EA, Wolfe K, Mumby PJ, Webster NS, Bourne DG (2020) Spatial patterns of microbial communities across surface waters of the Great Barrier Reef. Commun Biol 14:442. https://doi.org/10.1038/s42003-020-01166-y
Asif A, Mohsin H, Rehman Y (2021) Purple nonsulfur bacteria: An important versatile tool in biotechnology. Microb. Biotechnol:309–337. https://doi.org/10.1016/b978-0-12-822098-6.00003-3
Remple KL, Silbiger NJ, Quinlan ZA, Fox MD, Kelly LW, Donahue MJ, Nelson CE (2021) Coral reef biofilm bacterial diversity and successional trajectories are structured by reef benthic organisms and shift under chronic nutrient enrichment. NPJ Biofilms Microbiomes 7:84. https://doi.org/10.1038/s41522-021-00252-1
Oh HM, Kwon KK, Kang I, Kang SG, Lee JH, Kim SJ, Cho JC (2010) Complete genome sequence of “Candidatus Puniceispirillum marinum” IMCC1322, a representative of the SAR116 clade in the Alphaproteobacteria. J Bacteriol 192:3240–3241. https://doi.org/10.1128/JB.00347-10
Roda-Garcia JJ, Haro-Moreno JM, Huschet LA, Rodriguez-Valera F, López-Pérez M (2021) Phylogenomics of SAR116 Clade Reveals Two Subclades with Different Evolutionary Trajectories and an Important Role in the Ocean Sulfur Cycle. MSystems 6(5). https://doi.org/10.1128/msystems.00944-21
Kelly JB, Carlson DE, Low JS, Rice T, Thacker RW (2021) The relationship between microbiomes and selective regimes in the sponge genus Ircinia. Front Microbiol 11:607289. https://doi.org/10.3389/fmicb.2021.607289
Zhou J, Bai X, Zhao R (2017) Microbial communities in the native habitats of Agaricus sinodeliciosus from Xinjiang Province revealed by amplicon sequencing. Sci Rep 7:15719. https://doi.org/10.1038/s41598-017-16082-1
Hördt A, López MG, Meier-Kolthoff JP, Schleuning M, Weinhold LM, Tindall BJ, Gronow S, Kyrpides NC, Woyke T, Göker M (2020) Analysis of 1,000+ type-strain genomes substantially improves taxonomic classification of Alphaproteobacteria. Front Microbiol 11:468. https://doi.org/10.3389/fmicb.2020.00468
Krzmarzick MJ, Crary BB, Harding JJ, Oyerinde OO, Leri AC, Myneni SC, Novak PJ (2012) Natural niche for organohalide-respiring Chloroflexi. Appl Environ Microbiol 78:393–401. https://doi.org/10.1128/AEM.06510-11
Inagaki F, Hinrichs KU, Kubo Y, Bowles MW, Heuer VB, Hong WL, Hoshino T, Ijiri A, Imachi H, Ito M, Kaneko M, Lever MA, Lin YS, Methé BA, Morita S, Morono Y, Tanikawa W, Bihan M, Bowden SA et al (2015) DEEP BIOSPHERE. Exploring deep microbial life in coal-bearing sediment down to ~2.5 km below the ocean floor. Sci 24:420–424. https://doi.org/10.1126/science.aaa6882
Ahn YB, Rhee SK, Fennell DE, Kerkhof LJ, Hentschel U, Häggblom MM (2003) Reductive dehalogenation of brominated phenolic compounds by microorganisms associated with the marine sponge Aplysina aerophoba. Appl Environ Microbiol 69:4159–4166. https://doi.org/10.1128/AEM.69.7.4159-4166.2003
Kelly JB, Carlson DE, Low JS, Thacker RW (2022) Novel trends of genome evolution in highly complex tropical sponge microbiomes. Microbiome 4:164. https://doi.org/10.1186/s40168-022-01359-z
Sacristán-Soriano O, Banaigs B, Casamayor EO, Becerro MA (2011) Exploring the links between natural products and bacterial assemblages in the sponge Aplysina aerophoba. Appl Environ Microbiol 77:862–870. https://doi.org/10.1128/AEM.00100-10
Hug LA, Maphosa F, Leys D, Löffler FE, Smidt H, Edwards EA, Adrian L (2013) Overview of organohalide-respiring bacteria and a proposal for a classification system for reductive dehalogenases. Philos Trans R Soc Lond B Biol Sci 11:20120322. https://doi.org/10.1098/rstb.2012.0322
Busch K, Slaby BM, Bach W, Boetius A, Clefsen I, Colaço A, Creemers M, Cristobo J, Federwisch L, Franke A, Gavriilidou A, Hethke A, Kenchington E, Mienis F, Mills S, Riesgo A, Ríos P, Roberts EM, Sipkema D et al (2022) Biodiversity, environmental drivers, and sustainability of the global deep-sea sponge microbiome. Nat Commun 2:5160. https://doi.org/10.1038/s41467-022-32684-4
Anantharaman K, Brown CT, Hug LA, Sharon I, Castelle CJ, Probst AJ, Thomas BC, Singh A, Wilkins MJ, Karaoz U, Brodie EL, Williams KH, Hubbard SS, Banfield JF (2016) Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nature. Comm 7(1):13219. https://doi.org/10.1038/ncomms13219
Hiraoka S, Hirai M, Matsui Y, Makabe A, Minegishi H, Tsuda M, Juliarni RE, Danovaro R, Corinaldesi C, Kitahashi T, Tasumi E, Nishizawa M, Takai K, Nomaki H, Nunoura T (2020) Microbial community and geochemical analyses of trans-trench sediments for understanding the roles of hadal environments. ISME J 14:740–756. https://doi.org/10.1038/s41396-019-0564-z
Lopez-Fernandez M, Simone D, Wu X, Soler L, Nilsson E, Holmfeldt K, Lantz H, Bertilsson S, Dopson M (2018) Metatranscriptomes reveal that all three domains of life are active but are dominated by bacteria in the Fennoscandian crystalline granitic continental deep biosphere. mBio 9(6):10-1128. https://doi.org/10.1128/mBio.01792-18
Anantharaman K, Hausmann B, Jungbluth SP, Kantor RS, Lavy A, Warren LA, Rappé MS, Pester M, Loy A, Thomas BC, Banfield JF (2018) Expanded diversity of microbial groups that shape the dissimilatory sulfur cycle. ISME J 12:1715–1728. https://doi.org/10.1038/s41396-018-0078-0
Dalcin Martins P, Danczak RE, Roux S, Frank J, Borton MA, Wolfe RA, Burris MN, Wilkins MJ (2018) Viral and metabolic controls on high rates of microbial sulfur and carbon cycling in wetland ecosystems. Microbiome 7:138. https://doi.org/10.1186/s40168-018-0522-4
Löffler M, Wallerang KB, Venceslau SS, Pereira IAC, Dahl C (2020) The iron-sulfur flavoprotein DsrL as NAD(P)H:acceptor oxidoreductase in oxidative and reductive dissimilatory sulfur metabolism. Front Microbiol 11:578209. https://doi.org/10.3389/fmicb.2020.578209
O’Brien PA, Andreakis N, Tan S, Miller DJ, Webster NS, Zhang G, Bourne DG (2021) Testing cophylogeny between coral reef invertebrates and their bacterial and archaeal symbionts. Mol Ecol 30:3768–3782. https://doi.org/10.1111/mec.16006
Díez-Vives C, Taboada S, Leiva C, Busch K, Hentschel U, Riesgo A (2020) On the way to specificity - microbiome reflects sponge genetic cluster primarily in highly structured populations. Mol Ecol 29:4412–4427. https://doi.org/10.1111/mec.15635
Moitinho-Silva L, Bayer K, Cannistraci CV, Giles EC, Ryu T, Seridi L, Ravasi T, Hentschel U (2014) Specificity and transcriptional activity of microbiota associated with low and high microbial abundance sponges from the Red Sea. Mol Ecol 23:1348–1363. https://doi.org/10.1111/mec.12365
Neulinger SC, Stöhr R, Thiel V, Schmaljohann R, Imhoff JF (2010) New phylogenetic lineages of the Spirochaetes phylum associated with Clathrina species (Porifera). J Microbiol 48:411–418. https://doi.org/10.1007/s12275-010-0017-x
Walmsley TA, Matcher GF, Zhang F, Hill RT, Davies-Coleman MT, Dorrington RA (2012) Diversity of bacterial communities associated with the Indian Ocean sponge Tsitsikamma favus that contains the bioactive pyrroloiminoquinones, tsitsikammamine A and B. Mar Biotechnol 14:681–691. https://doi.org/10.1007/s10126-012-9430-y
Matcher GF, Waterworth SC, Walmsley TA, Matsatsa T, Parker-Nance S, Davies-Coleman MT, Dorrington RA (2017) Keeping it in the family: coevolution of latrunculid sponges and their dominant bacterial symbionts. Microbiologyopen 6:e00417. https://doi.org/10.1002/mbo3.417
Lilburn TG, Kim KS, Ostrom NE, Byzek KR, Leadbetter JR, Breznak JA (2001) Nitrogen fixation by symbiotic and free-living spirochetes. Science 29:2495–2498. https://doi.org/10.1126/science.1060281
Lim SJ, Davis BG, Gill DE, Walton J, Nachman E, Engel AS, Anderson LC, Campbell BJ (2019) Taxonomic and functional heterogeneity of the gill microbiome in a symbiotic coastal mangrove lucinid species. ISME J 13:902–920. https://doi.org/10.1038/s41396-018-0318-3
Ruiz C, Villegas-Plazas M, Thomas OP, Junca H, Pérez T (2020) Specialized microbiome of the cave-dwelling sponge Plakina kanaky (Porifera, Homoscleromorpha). FEMS Microbiol Ecol 96:fiaa043. https://doi.org/10.1093/femsec/fiaa043
Mori T, Cahn JKB, Wilson MC, Meoded RA, Wiebach V, Martinez AFC, Helfrich EJN, Albersmeier A, Wibberg D, Dätwyler S, Keren R, Lavy A, Rückert C, Ilan M, Kalinowski J, Matsunaga S, Takeyama H, Piel J (2018) Single-bacterial genomics validates rich and varied specialized metabolism of uncultivated Entotheonella sponge symbionts. Proc Natl Acad Sci USA 115:1718–1723. https://doi.org/10.1073/pnas.1715496115
Fieseler L, Horn M, Wagner M, Hentschel U (2004) Discovery of the novel candidate phylum “Poribacteria” in marine sponges. Appl Environ Microbiol 70:3724–3732. https://doi.org/10.1128/AEM.70.6.3724-3732.2004
Campos AB, Cavalcante LC, de Azevedo AR, Loiola M, Silva AET, Ara A, Meirelles PM (2022) CPR and DPANN have an overlooked role in corals’ microbial community structure. Microb Ecol 83:252–255. https://doi.org/10.1007/s00248-021-01737-4
González-Castillo A, Carballo JL, Bautista-Guerrero E (2021) Genomics and phylogeny of the proposed phylum ‘Candidatus Poribacteria’ associated with the excavating sponge Thoosa mismalolli. Antonie Van Leeuwenhoek 114:2163–2174. https://doi.org/10.1007/s10482-021-01670-z
Keller-Costa T, Lago-Lestón A, Saraiva JP, Toscan R, Silva SG, Gonçalves J, Cox CJ, Kyrpides N, Nunes da Rocha U, Costa R (2021) Metagenomic insights into the taxonomy, function, and dysbiosis of prokaryotic communities in octocorals. Microbiome 25:72. https://doi.org/10.1186/s40168-021-01031-y
Robbins SJ, Singleton CM, Chan CX, Messer LF, Geers AU, Ying H, Baker A, Bell SC, Morrow KM, Ragan MA, Miller DJ, Forêt S, Ball E, Beeden R, Berumen M, Aranda M, Ravasi T, Bongaerts P, Hoegh-Guldberg O (2019) A genomic view of the reef-building coral Porites lutea and its microbial symbionts. Nat. Microbiol 4(12):2090–2100. https://doi.org/10.1038/s41564-019-0532-4
Schmitt S, Tsai P, Bell J, Fromont J, Ilan M, Lindquist N, Perez T, Rodrigo A, Schupp PJ, Vacelet J, Webster N, Hentschel U, Taylor MW (2012) Assessing the complex sponge microbiota: core, variable and species-specific bacterial communities in marine sponges. ISME J 6:564–576. https://doi.org/10.1038/ismej.2011.116
Simister RL, Deines P, Botté ES, Webster NS, Taylor MW (2011) Sponge-specific clusters revisited: a comprehensive phylogeny of sponge-associated microorganisms. Environ Microbiol 14:517–524. https://doi.org/10.1111/j.1462-2920.2011.02664.x
de Menezes TA, de Freitas MAM, Lima MS, Soares AC, Leal C, Busch M, De S, Tschoeke DA, De O, Vidal L, Atella GC, Kruger RH, Setubal J, Vasconcelos AA, de Mahiques MM, Siegle E, Asp NE, Cosenza C, Hajdu E et al (2022) Fluxes of the Amazon River plume nutrients and microbes into marine sponges. Science of The Total Environment 847:157474. https://doi.org/10.1016/j.scitotenv.2022.157474
Lee SD (2019) Sneathiella limimaris sp. nov., a marine alphaproteobacterium isolated from a tidal mudflat and emended description of the genus Sneathiella. Int J Syst Evol Microbiol 69:1993–1997. https://doi.org/10.1099/ijsem.0.003411
Jordan EM, Thompson FL, Zhang XH, Li Y, Vancanneyt M, Kroppenstedt RM, Priest FG, Austin B (2007) Sneathiella chinensis gen. nov., sp. nov., a novel marine alphaproteobacterium isolated from coastal sediment in Qingdao. China. Int J Syst Evol Microbiol 57:114–121. https://doi.org/10.1099/ijs.0.64478-0
Khan SA, Jung HS, Park HY, Jeon CO (2021) Maritimibacter harenae sp. nov. and Sneathiella litorea sp. nov.: members of Alphaproteobacteria isolated from sea sand. Antonie Van Leeuwenhoek 114:799–811. https://doi.org/10.1007/s10482-021-01559-x
Pajares S, Varona-Cordero F, Hernández-Becerril DU (2020) Spatial distribution patterns of bacterioplankton in the oxygen minimum zone of the tropical Mexican Pacific. Microb Ecol 80:519–536. https://doi.org/10.1007/s00248-020-01508-7
Kurahashi M, Fukunaga Y, Harayama S, Yokota A (2008) Sneathiella glossodoripedis sp. nov., a marine alphaproteobacterium isolated from the nudibranch Glossodoris cincta, and proposal of Sneathiellales ord. nov. and Sneathiellaceae fam. nov. Int J Syst Evol Microbiol 58:548–552. https://doi.org/10.1099/ijs.0.65328-0
Quigley KM, Alvarez Roa C, Torda G, Bourne DG, Willis BL (2020) Co-dynamics of Symbiodiniaceae and bacterial populations during the first year of symbiosis with Acropora tenuis juveniles. Microbiologyopen 9:e959. https://doi.org/10.1002/mbo3.959
Sauret C, Séverin T, Vétion G, Guigue C, Goutx M, Pujo-Pay M, Conan P, Fagervold SK, Ghiglione JF (2014) ‘Rare biosphere’ bacteria as key phenanthrene degraders in coastal seawaters. Environ Pollut 194:246–253. https://doi.org/10.1016/j.envpol.2014.07.024
Muck S, De Corte D, Clifford EL, Bayer B, Herndl GJ, Sintes E (2019) Niche differentiation of aerobic and anaerobic ammonia oxidizers in a high latitude deep oxygen minimum zone. Front Microbiol 10:2141. https://doi.org/10.3389/fmicb.2019.02141
Acknowledgements
The specimens in Martinique were collected during the Madibenthos expeditions (PI Philippe Bouchet) organized by the Museum National d’Histoire Naturelle (MNHN) and the Marine Protected Areas Agency (AAMP), the Regional Directorate for the Environment (DEAL), and the Martinique Water Bureau (ODE). We would like to thank all participants for facilitating the fieldwork and making it an enjoyable experience.
Funding
Open access funding provided by FCT|FCCN (b-on). Funding was provided by the European Regional Development Fund (ERDF), the Territorial Collectivity of Martinique (CTM), and Saint-James Plantations and BRED Banque populaire. This work was supported by European Funds through COMPETE [FCOMP-01-0124-FEDER-008657] and by National Funds through the Portuguese Foundation for Science and Technology (FCT) within the LESS CORAL [PTDC/AAC-AMB/115304/2009] and Ecotech-Sponge (PTDC/BIAMIC/6473/2014 – POCI-01-0145-FEDER-016531) projects. We acknowledge financial support to CESAM from FCT/MCTES (UIDP/50017/2020+ UIDB/50017/2020 + LA/P/0094/2020), through national funds. FJRCC was funded by national funds through FCT – Fundação para a Ciência e a Tecnologia, I.P., under the Scientific Employment Stimulus - Individual Call – reference CEECIND/00070/2017. TMS was supported through 4D-REEF, a European Union's Horizon 2020 research and innovation programme, under the Marie Skłodowska-Curie grant agreement No 813360. ARMP was supported by a postdoctoral scholarship (SFRH/BPD/117563/2016) funded by the Portuguese Foundation for Science and Technology (FCT)/national funds (MCTES) and by the European Social Fund (ESF)/EU. VO was funded by National funds (OE), through FCT, IP., in the scope of the framework contract foreseen in the numbers 4, 5, and 6 of the Article 23, of the Decree-Law 57/2016, of August 29, changed by Law 57/2017, of July 19.
Author information
Authors and Affiliations
Contributions
All authors contributed to writing the manuscript. D. F. R. C. and F. J. R. C. C. analyzed the data and prepared the figures. T. S. contributed to the lab work. N. J. d. V. contributed to the fieldwork and identification of sponge specimens.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Supplementary Information
Supplementary data 1.
List of samples used in the present study including the sample-id, sample code (Code), group code (Group), Group_full, Species, sampling date (Date), sampling site (Site), Country, Depth (Dep), Latitude (Lat), Longitude (Lon), Country, and Naturalis museum registration number (RMNH POR). (XLS 19 kb)
Supplementary data 2.
OTU counts table including taxonomic assignment. (XLS 5426 kb)
Supplementary data 3.
List of the 50 most abundant OTUs recorded in the present report and their taxonomic assignment using the Silva (https://www.arb-silva.de/) database. (XLS 12 kb)
Supplementary data 4.
Results of emmeans analysis showing pairwise comparisons of differences in selected diversity indices between pairs of groups. (XLSX 11 kb)
Supplementary data 5.
Results of emmeans analysis showing pairwise comparisons of differences in the relative abundances of the six most abundant phyla between pairs of groups. (XLSX 15 kb)
Supplementary data 6.
Results of emmeans analysis showing pairwise comparisons of differences in the relative abundances of the six most abundant orders between pairs of groups. (XLSX 17 kb)
Supplementary data 7.
Ordination showing the third and fourth axes of the principal coordinates analysis (PCO) of OTU composition. Symbols are color coded and represent samples from different groups as shown in the legend on the right side of the figure. Gray symbols represent weighted averages scores for OTUs. The eigenvalues for the third and fourth axes were 0.73 and 0.38, respectively, and explained 6.3 and 3.3%, respectively, of the total variation in the data. The symbol sizes for OTUs are proportional to their abundances (number of sequence reads). The symbols refer to: Sediment (Sd), Water (Wt), Cinachyrella alloclada, (Ca), Cinachyrella kuekenthali in dimly lit (Cd) and illuminated (Cl) habitats, and Xestospongia muta sampled in dimly lit (Xd) and illuminated (Xl) habitats. (PDF 345 kb)
Supplementary data 8.
Boxplots showing the relative abundances of HMA indicator taxa. The x-axis labels refer to: Sediment (Sd), Water (Wt), Cinachyrella alloclada, (Ca), Cinachyrella kuekenthali in dimly lit (Cd) and illuminated (Cl) habitats, and Xestospongia muta sampled in dimly lit (Xd) and illuminated (Xl) habitats. Colored symbols indicate specimens collected from caves (Cv), deep (Dp), and shallow water (Sh). (PDF 10 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cleary, D.F.R., de Voogd, N.J., Stuij, T.M. et al. A Study of Sponge Symbionts from Different Light Habitats. Microb Ecol 86, 2819–2837 (2023). https://doi.org/10.1007/s00248-023-02267-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-023-02267-x