
Abstract
The population diversity of cultured isolates of the phylum Bacteroidetes was investigated from salt-marsh sediments. A total of 44 isolates that belonged to this phylum were isolated either from high-dilution plates or from end-dilution most-probable-number (MPN) tubes. The majority of the isolates came from Virginia, with others isolated from salt marshes in Delaware and North Carolina. All the isolates were aerobic Gram-negative, catalase positive small rods that formed uniform colonies; most had either yellow or orange pigmentation. Riboprinting of 40 isolates revealed they were genotypically diverse, consisting of 33 different riboprint patterns; there were four riboprint groups with two or more members. The isolates could be divided into 23 different fatty acid methyl ester (FAME) profiles at the species level with 14 of the profiles being unique to single isolates. One group of 10 isolates was closely related, suggesting this group may be well adapted for life in salt marshes. Thirteen of the isolates were selected for sequencing of the small-subunit ribosomal RNA gene representing a diverse group of isolates that fell within the classes Sphingobacteria and Flavobacteria. Only one of the isolates was >97% similar at the 16S rDNA to a described species of Cytophaga marinoflava; the other isolates were 94 to 96.5% related to undescribed isolates mostly within the class Flavobacteria. There was good concordance between the FAME dendrogram and a phylogenetic tree based on comparison of 16S sequences. There were no obvious temporal or spatial distribution patterns to the isolates, suggesting that this group of bacteria is inherently diverse.
Similar content being viewed by others

Introduction
The phylum Bacteroidetes was recently created [12] and incorporates members of the Cytophaga–Flavobacteria/Flexibacter–Bacteroides (CFB) division [27] into a taxonomically coherent group. The phylum is made up of three classes: the Bacteroidetes, the Flavobacteria, and the Sphingobacteria [12]. This last class includes the genera Cytophaga and Flexibacter. The class Bacteroidetes are, for the most part, strict anaerobes often associated with the intestinal tracts of animals. The classes Flavobacteria and Sphingobacteria represent a physiologically broad group of heterotrophic bacteria that are ubiquitous, occupying freshwater and marine habitats, as well as sediments and soils. Although there are no major metabolic characteristics that distinguish these latter two classes, many species within them are known for their ability to degrade complex polymers. This trait makes them important in the breakdown of lignocellulosic plant materials and, in part, explains their common abundance in terrestrial and aquatic ecosystems.
Analysis of a wide range of aquatic [11, 16, 19, 26, 37] and terrestrial ecosystems [3, 10, 17, 25] typically reveals that members of the CFB (as they have traditionally been referred to) may constitute between 5 and 20% of the clones from a given environment. In a recent review, Kirchman pointed out that microscopic counts of seawater samples using group specific fluorescent in-situ hybridization (FISH) probes has revealed that the Bacteroidetes are the most abundant single phylum enumerated in several analyses of open ocean waters [19]. In the studies mentioned above, the Bacteroidetes phylotypes that have been identified either from the clone libraries or from sequence analysis of denaturing gradient gel electrophoresis (DGGE) amplicons are not often closely related to known cultivated species of Bacteroidetes. This suggests at least two possibilities: (1) that many of the important environmental clones are especially difficult to cultivate in the laboratory; or (2) that there is a high background diversity of phylotypes within this group that results in the routine discovery of new lineages.
Although we are beginning to appreciate the abundance and diversity of marine Bacteroidetes populations in the open ocean and in coastal waters, there is little information on the presence and diversity of members of this phylum in salt marshes and more specifically salt-marsh sediments. Burke et al. [7] conducted a study of sediments associated with different plant types, Spartina patens and Phragmites australis, in a mesohaline marsh on the Hudson River. They used group specific fluorescence in situ hybridization (FISH) probes for a number of groups including the CFB319a probe and found Bacteroidetes were consistently present throughout the growth season; however, their numbers accounted for <1% of the total bacterial population. In investigations of diazotrophic bacteria associated with the rhizosphere of salt-marsh plants in South Carolina, Bagwell and colleagues [1] found that members of the Flavobacteria were present, but not in high numbers relative to other aerobic heterotrophs.
As part of a survey of different microbial groups associated with salt-marsh sediments, we investigated the dominant aerobic heterotrophic bacteria that could be cultivated and found that members of the Bacteroidetes were consistently present and in moderate abundance. The specific purpose of this study was to use genotypic and phenotypic methods to assess the population diversity of these cultured isolates and determine their inherent diversity as well as assess obvious spatial or temporal patterns to their distribution.
Materials and Methods
Sites and Sampling
Most of the isolates used for this analysis were collected from the Virginia Coastal Reserve (VCR), located on the eastern shore of Virginia as part of a larger survey of microbial diversity at this habitat. This site is part of a Long Term Ecosystem Research (LTER) project that is managed by the University of Virginia; details about the site are available at their Web site (http://atlantic.evsc.virginia.edu/). Samples were collected at all times of year (Table 1); however during one specific sampling trip in May 2000 (trip BSD), we focused our attention on collecting members of the phylum Bacteroidetes. The primary sampling sites were Hog Island S1 and S2, Cattleshed marsh (CS), and Red Bank (RB). Hog Island, located 15 km from the mainland, is a pristine barrier island and the S1 and S2 sites were located near the upland margin of a Spartina alterniflora–dominated marsh (37° 27.167′ N; 075° 40.513′ W). The S1 site was in a periodically flooded stand of tall-form S. alterniflora, and the sediment was a fine silt that was gray-black. The S2 site was ~3 m from the S1 site in a small open area devoid of growing S. alterniflora that was often submerged, and the sediment was composed of a black mud rich in organic matter. The CS site was in a large marsh of tall-form S. alterniflora ~1.5 km from Hog Island (37° 26.595′ N; 075° 41.315′ W), and the sediment was composed of a gray mixture of fine silt/clay. Red Bank was on the bank of a creek located within 100 m of the mainland and 15 km from Hog Island (7° 26.731′ N; 075° 50.382′ W); the sediment was gray and had a high clay content. Samples were also collected from three other sites. Oyster Creek (OC) near Oyster, VA (~50 km from Hog Island) was in tall-form S. alterniflora in a small marsh that bordered an upland forest. The samples from Delaware (DE) were taken in short-form S. altneriflora from the Great Marsh, which is a large barrier marsh near Lewes. The samples from North Carolina (NC) were taken in tall-form S. alterniflora from marshes near Morehead City that were located along the intercoastal waterway. At all these sites the salinities were in the range of 30–33 ppt.
At all the sites 15-cm-deep sediment cores were collected using 3-inch-diameter PVC pipe corers. The core barrels were sterilized by rinsing them first with a 10% bleach solution and then with sterilized deionized water. The cores were stored on ice and returned to the laboratory where they were processed within 24 h. A slurry was made from the top 5–7 cm of the sediment by diluting it with a small amount of sterile seawater. This slurry was used to quantitate the numbers of aerobic, heterotrophic bacteria that could be cultivated, either by dilution plating onto R2A medium (Difco, Detroit. MI) containing 2% NaCl, or by inoculation of most-probable-number (MPN) tubes also containing R2 broth with 2% NaCl. When MPNs were used, samples from the highest dilution tubes that had growth were streaked onto R2A plates for isolation and selection of isolates as described below.
Selection of Isolates
Isolated colony types characteristic of Bacteroidetes, small and round with carotenoid-type pigmentation, were selected from the initial R2A isolation plates and streaked on fresh plates. This procedure was repeated three times, each time picking well isolated colonies to obtain axenic cultures. The selected isolates were examined microscopically and analyzed by FAME (see below) to determine if they had fatty acid profiles characteristic of the Bacteroidetes. Further characterization was carried out as described below, and stocks of each isolate were stored in liquid nitrogen using 10% (w/v) glycerol (final concentration) as a preservative. Type strains were from the American Type Culture Collection.
Biochemical Tests
All of the isolates were tested for Gram reaction, motility, catalase and oxidase activities, growth at temperatures between 12°C and 37°C, and the presence of flexirubins. Gram-staining and catalase tests were done using standard methods [13]. Motility was observed under a wet-mount preparation. Oxidase activity was tested using oxidase DrySlide reagent from BBL and looking for the formation of a dark blue pigment. The presence of flexirubins was tested by adding a solution of 10% KOH to colonies on filter paper and looking for the appearance of a red pigment.
Fatty Acid Methyl Ester (FAME) Analysis
Each strain was streaked onto R2A agar and incubated at 22–26°C. After confluent growth was obtained, which usually took between 72 and 96 h, 40–50 mg of cells was harvested from the plate. The fatty acid methyl esters were extracted for taxonomic purposes [33] according to the standardized protocol of the Microbial Identification System (MIDI) (MIDI Inc., Newark, DE). The extracted samples were analyzed with a Hewlett-Packard HP6890 gas chromatograph and the profiles were compared with the Sherlock TSBA library 4.0 version (Microbial ID, MIDI Inc.) A Euclidean dendrogram comparing all the isolates was created using the MIDI software.
Genotyping by Riboprint
The RiboPrinter (Qualicon, Wilmington, DE) microbial characterization system is a robotic molecular biology workstation [6] that automatically carries out a restriction digest of the chromosomal DNA and performs a Southern blot using a bacterial probe based on the ribosomal DNA operon. The result is a DNA fingerprint which is strain specific. For this study, the strains were grown on R2A medium for 72–96 h. Individual colonies were picked and homogenized into a lysing buffer, and then loaded into the machine, which automates the restriction digestion, gel electrophoresis, and Southern blot. The restriction enzyme EcoRI was used for these analyses. The riboprinter uses image processing software to generate a riboprint from the gel blot. The riboprints were exported to Bionumerics software (Applied Maths, Kortjik, Belgium) for comparison and creation of dendrograms. The dendrograms were constructed using the unweighted pair-group algorithm with averages (UPGMA) and the Pearson product-moment correlation coefficient as implemented by Bionumerics.
DNA Extraction and 16S rRNA Analysis
Genomic DNA was extracted from cell pellets using slight modifications to the FastDNA SPIN Kit for Soil (BIO 101, Vista, CA). Samples were placed in the kit’s MULTIMIX 2 tubes with the appropriate buffers and reagents, placed in the FastPrep instrument (BIO 101), and processed for 20 s at a setting of 5.5. The manufacturer’s protocol was then followed with DNA elution in 65°C DEPC water.
Amplification of the entire 16S rRNA gene was accomplished using primers L27F and L1492R [20]. Primer numbering is in reference to the alignment with Escherichia coli. To ensure accurate sequence data, the 16S rRNA gene was sequenced using 16 primers producing redundant double-stranded coverage. Amplification was performed using a PTC thermal cycler (MJR Research) and Ampli-Taq Gold with the GeneAmp 10× PCR Buffer II Kit (PerkinElmer Corp.). The reaction mixture of 100 μL total volume consisted of 10 mM Tris-HCl, 50 mM KCl, 0.01% gelatin, 2.5 mM MgCl2, 0.25 mM dNTPs, 0.5 μM of each primer, 1 ng/μL genomic DNA and 0.0025 U/μL of TaqGold DNA polymerase. Preincubation to activate the TaqGold polymerase was performed at 95°C for 10 min. The samples were subjected to 30 touchdown cycles (decreasing 2°C every two cycles from 65°C to 55°C) and were run under the following conditions: denaturing at 94°C for 1 min, annealing for 1 min at 55°C, and extension at 72°C for 2 min. After the last cycle, the samples were incubated at 72° for 10 min. A 5-μL aliquot of the reaction mixture was analyzed by electrophoresis (1% agarose), and the reaction products were visualized under UV light after staining with ethidium bromide.
DNA Sequencing
The PCR products for cycle sequencing were purified using the Wizard PCR DNA Purification System (Promega, Madison, WI) according to the supplier’s instructions. The sequencing reactions were carried out with a Taq DyeDeoxy Terminator Cycle Sequencing Kit (Applied Biosystems) and purified on Bio-Spin chromatography columns containing Sephadex G-50 (Bio-Rad Laboratories). The dried samples were dissolved in loading buffer containing formamide and were run on a 377 DNA Sequencer following the standard thermal cycle sequencing protocol (Applied Biosystems). The 16S rRNA gene sequence fragments for each strain were aligned using Sequencher (Gene Codes Corp.), single-stranded ends were trimmed off after alignment, and the consensus sequence for each strain was determined. The consensus sequence of each strain had at least twofold coverage in each direction.
Data Analysis
The consensus sequences from all strains were aligned using CLUSTALW [35] and the alignment of signature sequences was optimized manually using the SEQLAB multiple sequence editor [31] in the GCG software package distributed by Accelrys Inc. The multiple sequence alignments were trimmed, and the resulting data matrix encompassed 11,523 base pairs. A maximum likelihood tree was constructed utilizing the heuristic search option in PAUP [34] with the HKY85 substitution model (kappa = 3.885), the Newton–Raphson algorithm for branch length optimization, and the tree-bisection–reconnection algorithm for branch swapping. A bootstrap analysis was performed using 100 samplings, and the resulting bootstrap values were placed on the corresponding branches of the maximum likelihood tree. All the sequences have been placed in GenBank with accession numbers AY259501–AY259513.
Results
General Characterization of Isolates
We collected a total of 44 isolates at different times of the year from three separate geographical locations on the east coast of the United States. The numbers of isolates are summarized in Table 1. As mentioned above, the May 2000 sampling trip to the VCR (BSD) was focused on collecting and characterizing isolates that were members of the phylum Bacteroidetes; thus these isolates accounted for 61% of all the strains. The other isolates were representative of strains obtained from other sampling trips at the VCR, or from sampling trips to Delaware and North Carolina and were confirmed to be members of the Bacteroidetes. The selected strains were all from the highest dilution plates (10−5) or MPN tubes (10−7 –10−9) that had growth. All the strains shared similar phenotypic characteristics. They were all Gram-negative, short rods that formed small, round, pigmented colonies on plates; most were yellow and some were orange or pink. Figure 1 shows the typical cell morphology for several strains. None of the strains exhibited gliding motility under the growth conditions that were used; however, some of them did appear to swim. All the strains were catalase positive and oxidase positive. All of the BSD isolates were tested for the presence of flexirubins; however, only three of 25 strains contained these pigments that are considered indicative of the genus Cytophaga [29]. Some of the BSD isolates gave presumptive evidence for nitrate reduction; however, none showed fermentative growth in the absence of oxygen.

Photomicrographs of three different isolates of bacteria isolated during this study. These were from the CS and S2 sites at the VCR and represent widely divergent FAME types. All the isolated strains were short to medium-length rods, and there was some variability in the cell diameters. The bars equal 10 μm.
RiboPrint Analysis
The RiboPrint dendrogram shown in Fig. 2 illustrates the substantial genotypic variability that existed among these isolates. Using the benchmark that Riboprints with ≥90% similarity are the same genotype, we observed that 33 of the 40 (83%) unknown isolates that were tested had distinct genotypes. Four patterns were exhibited by more than one isolate. Cluster IV had one member from the CS site and three from the S1 site on Hog Island. Cluster III had one member from the Hog Is S1 site and 2 from the Hog Is S2 site; cluster II had two members from the Hog Island S2 site that were collected at different times (May 2000 and February 2001), and cluster I had two members from the CS site. None of the unknown Bacteroidetes isolates were identified by the RiboPrinter database, and as shown in Fig. 2, none were closely related to the described species in this phylum that were included in the analysis.

Dendrogram of RiboPrint data. The bold Roman numerals represent clusters of two or more RiboPrints that had ≥90% similarity. The named isolates are known members of the classes Flavobacteria and Sphingobacteria included for comparative purposes.
FAME Profile of CFB-like Isolates
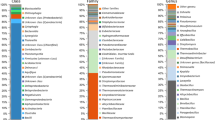
One of the defining characteristics of most of the members of the Bacteroidetes is that they contain 15:0 iso-branched and 17:0 iso-branched-chain fatty acids, which are considered a signature for this group [29]. We used FAME to screen our original isolates and determined those that belonged to the Bacteroidetes by the presence of the above characteristic fatty acids. Many of the strains that were analyzed also had iso-branched 3-OH groups at the 15th, 16th, and 17th carbon, which is another characteristic of this phylum [29]. The results of FAME analysis of 43 of the strains are shown in Fig. 3. None of the strains had a high identity to the members of the classes Flavobacteria or Sphingobacteria that are in the MIDI databank (11 strains). As is apparent, none of the strains appeared identical by FAME analysis; however, there were nine clusters of strains that had more than one member related at a Euclidean distance of ≤10, which is considered the level of species separation for FAME [15]. Using this value indicates there were 23 putative species represented among the 43 strains we sampled; 14 of these are represented by only one isolate. The FAME cluster A was by far the largest, with 10 representatives. The three types strains that we used as representatives of the class Flavobacteria, Zobellia uliginosa (ATCC 14397) and Flavobacterium johnsoniae (ATCC 17061), and the class Sphingobacteria, Cytophaga marinoflava (ATCC 19326), were not closely related to any of the new isolates.

Dendrogram of FAME data. The hatched line shows a Euclidean distance of 10; strains falling below this distance are considered to be in the same species, and strains above may be different species. The bold letters denote clusters of strains that are considered related at the species level. The strains listed in bold are those that also had their 16S genes sequenced. The named isolates are known members of the Flavobacteria and Sphingobacteria included for comparative purposes. The isolate BSB S2-06 was included as an outgroup and was identified as a member of the γ-Proteobacteria most closely related to Microbulbifer sp. by 16S rDNA analysis (results not shown). The complete FAME profiles for each organism are available online (http://Biocomplexity.binf.gmu.edu).
16 SSU rRNA Analysis
Based on the results of FAME analysis 13 strains were chosen for sequence comparison of their 16S rRNA genes. Four isolates (BSD S1-19, BSD S2-05, BSD S2-03, and BSD S1-18) were part of the closely related FAME cluster A, while the other nine strains were scattered through the FAME dendrogram. The phylogenetic tree is shown in Fig. 4. Overall, the isolates that were sequenced represent a wide phylogenetic diversity within the classes Flavobacteria and Sphingobacteria and appear to be concordant with the FAME dendrogram (Fig. 4, inset). The four isolates from the Group A in the FAME comparison were all very closely related to one another, as shown in Table 2. Using the benchmark that a similarity of ≥97% at the SSU rDNA sequence level indicates that two organisms belong to the same species [32]; the table shows that only BSD S1-22 is >97% similar to a described species, Cytophaga marinoflava. The other isolates had sequence similarities that ranged from 94 to 96.5% to other strains that by SSU rRNA sequence fall within the Bacteroidetes phylum, although none of these isolates are validly described taxa.

Phylogenetic tree based on 16S rDNA sequences showing the relationship of 13 strains from this study to other members of the Bacteroidetes. The tree was constructed using maximum likelihood. The nodes that had bootstrap values >50 are labeled; an asterisk denotes that the value for that particular branch was also >50. The inset shows the corresponding FAME dendrogram for these same strains. Note the overall concordance between these two trees.
The closest relatives to the strains analyzed by 16S phylogeny are all marine bacteria isolated from a wide variety of sources (Table 2). Cytophaga sp. ABO15262, ABO15261 [22], and ABO15545 [21] are isolates from the deep sea. Marine bacterium AB032514 was isolated from a marine sponge (GenBank accession only). Cytophaga sp. AB073589 was isolated in association with a green alga (GenBank accession only), and marine bacterium AF359540 was isolated in association with a dinoflagellate [18]. The isolate BSD S2 02 was most closely related to a newly described genus, Aequorivita antarctica [5].
Discussion
The results of this study suggest that there is substantial diversity for the cultured strains within the classes Flavobacteria and Sphingobacteria that were isolated from salt-marsh sediments. It should be emphasized that the same selection, i.e., sampling strategy and growth conditions, was applied to isolating all these strains, and the strains were selected in part because they had similar colony and cell morphologies. One might assume that had the selective conditions been varied more, e.g. different growth media or growth temperatures used, the diversity would be even higher. Although the majority of strains were not closely related to described genera or to one another, the FAME cluster A was a significant exception. The isolates in this cluster were 3 times more abundant than any other cluster, which suggests that these organisms may be particularly well adapted to living in the salt marsh. We have characterized one of these strains, BSD S2-05, further, and have proposed a new species, Gelidibacter salipaludis. sp. nov. [23]. Based on the four strains that were sequenced and the close FAME matches, as well as similar phenotypes, all 10 of the strains in this group will likely fall under this same species. G. algens was the first species described in this genus and was isolated from sea ice in Antarctica. It is a psychrophile that grows optimally between 15 and 18°C [4]. Another recently described species, G. mesophilus, was isolated from the Mediterranean Sea [24]. It grew at temperatures of up to 37°C.
Comparison of Genotypic, FAME, and 16S Sequence Diversity
There appears to be good concordance between the FAME dendrogram and the 16S phylogenetic tree in terms of differentiating between closely related strains and less related strains (Fig. 4). The FAME cluster A contains the 4 isolates that are >98% similar by SSU rRNA analysis. All the strains that fell below 97% similarity at the 16S level were also identified as different species by FAME. Although the RiboPrint data indicate that there is significant genotypic diversity, it is difficult to attribute any patterns to this data other than the obvious observation that some strains are essentially identical. It is interesting that three (II, III, and IV) of the four clusters with more than one RiboPrint type all fell within the FAME cluster A, although their RiboPrint patterns did not necessarily show high similarity to one another (Fig. 2). This further supports these organisms being closely related, though not all sharing the same genotype. The other FAME clusters did not show such a high degree of similarity by riboprinting.
Distribution Patterns of the Isolates
There did not appear to be any clear spatial or temporal patterns to the distribution of these isolates. Although the sampling is heavily biased toward the May 2000 sampling (BSD) of the four sites (S1, S2, CS, and RB), samples were collected from these same sites at different times (BSA, BSB, BSE, BSG, and BSM) and samples collected from Delaware (BSL) and North Carolina (BSJ) are interspersed throughout both the FAME and RiboPrint dendrograms. Cluster A from the FAME analysis was biased toward the Hog Island S1 and S2 sites, since eight of the 10 isolates were from this site. This is probably not surprising because these sites were located within 3 m of each other and several kilometers from the other sites. However, isolate BSD CS-27 was from Cattleshed, 1.5 km away, and isolate BSL D4-01 was collected from Delaware. In another example, cluster F from the FAME analysis yielded three related strains, BSA S1-04, BSG OC-02, and BSG RB-04, that were collected at different times and from three different VCR sites that were all several kilometers apart from one another. Of the nine clusters of related strains in the FAME results, only two, clusters E and I, had both strains isolated from the same site at the same time (BSD RB and S1, respectively).
Ecological Significance
To our knowledge, this is the first study that has specifically investigated the diversity of the Bacteroidetes in salt-marsh sediments. As mentioned above, other broader phylum studies have noted their presence, although they indicated that the Bacteroidetes were of minor numerical significance. We have also carried out extensive cultural studies at the same VCR sites described in this study using MPNs to quantitate different physiological groups such as S oxidizers, fermenters, sulfate-reducing bacteria, Fe-reducing bacteria, and S-dis proportionating bacteria, as well as aerobic heterotrophs. Based on these studies, aerobic heterotrophs were consistently the most abundant group recovered by MPN, with overall average numbers in the range of 108 cells g dry wt−1 sediment; this was ~10 times higher than the numbers of anaerobes that were found (Emerson, unpublished results). We have not precisely quantitated the numbers of the Bacteroidetes in all these samples, but based on this study, and the numbers of yellow-pigmented organisms recovered in general, we estimate that 5–20% of the recovered aerobic isolates obtained from these MPNs belong to the this phylum. Futhermore, examination of clone libraries containing more than 230 bacterial clones from the Hog Island S2 site and the CS site found that ~10% of the clones fell in the phylum Bacteroidetes, principally within the classes Sphingobacteria and Flavobacteria (Gillevet et al. manuscript in preparation). A comparison of the cultured strains that had 16S sequences (Fig. 4) and the Bacteroidetes environmental clones did not reveal any identities >97% similar between the clones and the pure cultures. The majority of clones clustered into clades that did not include members cultured during this study, and the clones themselves were diverse (results not shown). These results confirm that members of this phylum exhibit a good deal of diversity at the 16S rDNA level, and further suggest, perhaps not surprisingly, that the cultured organisms are not that well represented in the clone library.
It is not surprising that members of the classes Flavobacteria and Sphingobacteria would be common in an environment such as salt-marsh sediment that is rich in plant biomass. Given their metabolic propensity for the mineralization and/or degradation of both higher molecular weight dissolved organic matter and particulate plant material, they are thought to play an important role in catalyzing the detrital microbal food web that ultimately leads to the mineralization of organic matter [8, 19]. In a study that employed an artificially constructed, strictly anaerobic sediment microcosm, the Bacteroidetes showed the most significant population response to addition of complex organic matter, suggesting they could play a crucial role in providing low-MW organics for mineralization by sulfate-reducing bacteria [30]. The aerobic isolates tested in our study did not ferment, again suggesting that an even more diverse population of Bacteroidetes may be present in salt-marsh sediments than the diverse isolates described here.
Molecular-based population studies that have looked at the water column associated with either salt marshes or similar estuarine environments have found significant evidence for members of the Flavobacteria/Sphingobacteria classes in clone libraries as well as in cultured isolates. A comparison of two coastal lagoon systems in France, one oligotrophic and one eutrophic, found in both cases that the most abundant 16S clone types were members of the Bacteroidetes [2]. A study of the Columbia River estuary found that members of Bacteroidetes were among the most abundant clones recovered from particle-associated materials in the estuary itself. Interestingly, they were much less abundant on particulate material either from the nearby coastal ocean or in the freshwater portions of the Columbia River. The authors speculated that members of the Cytophaga group might be considered a “hallmark” of the estuarine environment [9]. Clearly, more studies focused on the relationship between sediment-associated and water-column-associated members of the Bacteroidetes within salt-marsh and estuarine environments would be enlightening.
Our results are also consistent with other cultivation-based investigations from the marine environment. One such study looked at a limited number of isolates from an enrichment that used salt-marsh water as the inoculum and found all of these were quite phylogenetically distinct from known species [14]. A substantial diversity of Bacteroidetes was also found in a study of culturable bacteria isolated from the water column of the North Sea using a variety of different growth conditions [36]. A similar study in the Baltic Sea indicated that members of this phylum were among the dominant groups that could be cultivated [28]. A more recent study that used a novel, environmentally relevant cultivation technique found an even greater diversity of Bacteroidetes in Sargasso Sea water [38], suggesting that this environmentally important but understudied phylum holds much promise as a source of novel diversity that can be cultivated and studied in the laboratory.
References
CE Bagwell YM Piceno A Ashburne-Lucas CF Lovell (1998) ArticleTitlePhysiological diversity of the rhizosphere diazotroph assemblages of selected salt marsh grasses. Appl Environ Microbiol 65 4276–4282
S Benlloch F Rodriquez-Valera AJ Martinez-Murcia (1995) ArticleTitleBacterial diversity in two coastal lagoons deduced from 16S rDNA PCR amplification and partial sequencing. FEMS Microbiol Ecol 18 267–280 Occurrence Handle10.1016/0168-6496(95)00067-4 Occurrence Handle1:CAS:528:DyaK2MXhtVSgtr3M
J Borneman PW Sckrock KM O’Sullivan JA Palus NG Rumjanek LL Jansen J Nienhuis EW Triplett (1996) ArticleTitleMolecular microbial diversity of an agricultural soil in Wisconsin. Appl Environ Microbiol 62 1935–1943 Occurrence Handle1:CAS:528:DyaK28XjtlGgsr0%3D Occurrence Handle8787391
J Bowman SA McCammon JL Brown PD Nichols TA McKeekin (1997) ArticleTitlePsychroserpen burtonensis gen. nov., sp. nov., and Gelidibacter algens gen. nov., pyschrophilic bacteria isolated from Antarctic lacustrine and sea ice habitats. Int J Syst Bacteriol 47 670–677 Occurrence Handle1:STN:280:ByiA2s%2FlvFY%3D Occurrence Handle9226898
JP Bowman DS Nichols (2002) ArticleTitleAequorivita gen. nov., a member of the family Flavobacteriaceae isolated from terrestrial and marine Antarctic habitats. Int J Syst Evol Microbiol 52 1533–1541 Occurrence Handle10.1099/ijs.0.01976-0 Occurrence Handle1:CAS:528:DC%2BD38XnvFyiur0%3D Occurrence Handle12361255
J Bruce (1996) ArticleTitleAutomated system rapidly identifies and characterizes microorganisms in food. Food Technol 50 77–81
DJ Burke EP Hamerlynck D Hahn (2002) ArticleTitleInteractions among plant species and microorganisms in salt marsh sediments. Appl Environ Microbiol 68 1157–1164 Occurrence Handle10.1128/AEM.68.3.1157-1164.2002 Occurrence Handle1:CAS:528:DC%2BD38XitV2qu7k%3D Occurrence Handle11872463
M Cottrell D Kirchman (2000) ArticleTitleNatural assemblages of marine proteobacteria and members of the Cytophaga–Flavobacter cluster consuming low- and high-molecular-weight dissolved organic matter. Appl Environ Microbiol 66 1692–1697 Occurrence Handle10.1128/AEM.66.4.1692-1697.2000 Occurrence Handle1:CAS:528:DC%2BD3cXisVWls7o%3D Occurrence Handle10742262
BC Crump EV Armburst JA Baross (1999) ArticleTitlePhylogenetic analysis of particle-attached and free-living bacterial communities in the Columbia River, its estuary, and the adjacent coastal ocean. Appl Environ Microbiol 66 3044–3051
J Dunbar SM Barns LO Ticknor CR Kuske (2002) ArticleTitleEmpirical and theoretical bacterial diversity in four Arizona soils. Appl Environ Microbiol 68 3035–3045 Occurrence Handle10.1128/AEM.68.6.3035-3045.2002 Occurrence Handle1:CAS:528:DC%2BD38XksVWqt7k%3D Occurrence Handle12039765
H Eilers J Pernthaler FO Glockner R Amann (2000) ArticleTitleCulturability and in situ abundance of pelagic bacteria from the North Sea. Appl Environ Microbiol 66 3044–3051 Occurrence Handle10.1128/AEM.66.7.3044-3051.2000 Occurrence Handle1:CAS:528:DC%2BD3cXkslKlt7k%3D Occurrence Handle10877804
Garrity, GM, Johnson, KL, Bell, JA, Searles, DB (2002) Taxonomic outline of the procaryotes. In: Bergey’s Manual of Systematic Bacteriology, 2nd ed, Springer-Verlag, New York
P Gerhardt RGE Murray WA Wood NR Krieg (1994) Methods for General and Molecular Bacteriology American Society for Microbiology Washington, DC
JM Gonzalez WB Whitman RE Hodson MA Moran (1996) ArticleTitleIdentifying numerically abundant culturable bacteria from complex communities: an example from a lignin enrichment culture. Appl Environ Microbiol 62 4433–4440 Occurrence Handle1:CAS:528:DyaK28Xnt1antLY%3D Occurrence Handle8953714
SK Haack H Garchow DA Odelson LJ Forney MJ Klug (1994) ArticleTitleAccuracy, reproducibility, and interpretation of fatty acid methyl ester profiles of model bacterial communities. Appl Environ Microbiol 60 2483–2493 Occurrence Handle1:CAS:528:DyaK2cXlt12jtrg%3D
A Hagstrom T Pommier F Rohwer K Simu W Stolte D Svensson UL Zweifel (2002) ArticleTitleUse of 16S ribosomal DNA for delineation of marine bacterioplankton species. Appl Environ Microbiol 68 3628–3633 Occurrence Handle10.1128/AEM.68.7.3628-3633.2002 Occurrence Handle12089052
U Hengstmann K Chin PH Janssen W Liesack (1999) ArticleTitleComparative phylogenetic assignment of environmental sequences of genes encoding 16S rRNA and numerically abundant culturable bacteria from anoxic rice paddy soil. Appl Environ Microbiol 65 5050–5058 Occurrence Handle1:CAS:528:DyaK1MXnt1Wmu7o%3D Occurrence Handle10543822
GL Hold EA Smith MS Rappe EW Maas ERB Moore C Stroempl JR Stephen JI Prosser TH Birkbeck S Gallacher (2001) ArticleTitleCharacterisation of bacterial communities associated with toxic and non-toxic dinoflagellates: Alexandrium spp. and Scrippsiella trochoidea. FEMS Microbiol Ecol 37 161–173 Occurrence Handle10.1016/S0168-6496(01)00157-X Occurrence Handle1:CAS:528:DC%2BD3MXnslCisbs%3D
D Kirchman (2002) ArticleTitleThe ecology of Cytophaga–Flavobacteria in aquatic environments. FEMS Microbiol Ecol 39 91–100 Occurrence Handle10.1016/S0168-6496(01)00206-9 Occurrence Handle1:CAS:528:DC%2BD38XitFaqt7k%3D
DJ Lane (1991) 16S/23S rRNA sequencing. E Stackebrandt M Goodfellow (Eds) Nucleic Acid Techniques in Bacterial Systematics John Wiley & Sons London 115–175
L Li C Kato K Horikoshi (1999) ArticleTitleBacterial diversity in deep-sea sediments from different depths. Biodivers Conserv 8 659–677 Occurrence Handle10.1023/A:1008848203739
L Li C Kato K Horikoshi (1999) ArticleTitleMicrobial diversity in sediments collected from the deepest cold-seep area, the Japan Trench. Mar Biotechnol 1 391–400 Occurrence Handle1:CAS:528:DyaK1MXmslKiurw%3D Occurrence Handle10489418
Lydell, C, Irby, A, Emerson, D (2003) Cyclobacterium virginiensis sp. nov., and Gelidibacter salipaludis sp. nov., two new members of the phylum Bacteroidetes isolated from coastal salt marshes. Abstr Ann Mtg Am Soc Microbiol, Washington, DC, p 616
MC Macian MJ Pujalte MC Marquez W Ludwig A Ventosa E Garay KH Schleifer (2002) ArticleTitleGelidibacter mesophilus sp. nov., a novel marine bacterium in the family Flavobacteriaceae. Int J Syst Evol Microbiol 52 1325–1329 Occurrence Handle10.1099/ijs.0.02009-0 Occurrence Handle1:CAS:528:DC%2BD38Xmt1GrsrY%3D Occurrence Handle12148647
AE McCaig L Glover JI Prosser (1999) ArticleTitleMolecular analysis of bacterial community structure and diversity in unimproved and improved upland grass pastures. Appl Environ Microbiol 65 1721–1730 Occurrence Handle1:CAS:528:DyaK1MXitlartbc%3D Occurrence Handle10103273
LA O’Sullivan AJ Weightman JC Fry (2002) ArticleTitleNew degenerate Cytophaga–Flexibacter–Bacteroides–specific 16S ribosomal DNA- targeted oligonucleotide probes reveal high bacterial diversity in River Taff epilithon. Appl Environ Microbiol 68 201–210 Occurrence Handle10.1128/AEM.68.1.201-210.2002 Occurrence Handle1:CAS:528:DC%2BD38Xjt1WnsQ%3D%3D Occurrence Handle11772628
BJ Paster W Ludwig WG Weisburg E Stackerbrandt RB Hespell CM Hahn H Reichenbach KO Stetter CR Woese (1985) ArticleTitleA phylogenetic grouping of the Bacteroides, Cytophagas, and certain Flavobacteria. Syst Appl Microbiol 6 32–42
J Pinhassi U Zweifel A Hagstrom (1997) ArticleTitleDominant marine bacterioplankton species found among colony-forming bacteria. Appl Environ Microbiol 63 3359–3366 Occurrence Handle1:CAS:528:DyaK2sXmtVKhsbc%3D Occurrence Handle9292985
H Reichenbach (1992) The order Cytophgagales. A Balows HG Trüper M Dworkin W Harder K-H Schleifer (Eds) The Prokaryotes Springer-Verlag New York 355–375
R Rosello-Mora B Thamdrup H Schafer R Weller R Amann (1999) ArticleTitleThe response of the microbial community of marine sediments to organic carbon input under anaerobic conditions. Syst Appl Microbiol 22 237–248 Occurrence Handle1:CAS:528:DyaK1MXkt1altLw%3D Occurrence Handle10390875
SW Smith R Overbeek CR Woese W Gilbert PM Gillevet (1994) ArticleTitleThe Genetic Data Environment (GDE): an expandable graphic interface for manipulating molecular information. CABIOS 10 671–675 Occurrence Handle1:CAS:528:DyaK2MXjvVKksLc%3D Occurrence Handle7704666
E Stackebrandt BM Goebel (1994) ArticleTitleA place for DNA–DNA reassociation and 16S ribosomal-RNA sequence-analysis in the present species definition in bacteriology. Int J Syst Bacteriol 44 846–849 Occurrence Handle1:CAS:528:DyaK2MXitFCnsb4%3D
K Suzuki M Goodfellow AG O’Donnell (1993) Cell envelopes and classification. M Goodfellow AG O’Donnell (Eds) Handbook of New Bacterial Systematics Academic Press London 195–250
DL Swofford (2001) PAUP* Phylogenetic Analysis Using Parsimony (*and Other Methods) Sinauer Associates Sunderland, MA
JD Thompson TJ Gibson F Plewniak F Jeanmougin DG Higgins (1997) ArticleTitleThe ClustalX Windows interface: flexible strategies for multiple sequence alignment aided by quality Analysis tools. Nucleic Acids Res 24 4876–4882 Occurrence Handle10.1093/nar/25.24.4876
HU Uphoff A Felske W Fehr I Wagner-Dobler (2001) ArticleTitleThe microbial diversity in picoplankton enrichment cultures: a molecular screening of isolates. FEMS Microbiol Ecol 35 249–258 Occurrence Handle10.1016/S0168-6496(01)00096-4 Occurrence Handle1:CAS:528:DC%2BD3MXislGrsb8%3D Occurrence Handle11311435
LE Wells JW Deming (2003) ArticleTitleAbundance of Bacteria, the Cytophaga–Flavobacterium, cluster, and Archaea in cold oligotrophic waters and neploid layers of the Northwest Passage, Canadian Archipelago. Aquat. Microb Ecol 31 19–31
K Zengler G Toledo M Rappe J Elkins EJ Mathur M Keller (2002) ArticleTitleCultivating the uncultured. Proc Natl Acad Sci USA 99 15681–15686
Acknowledgments
We thank the boat captains at the VCR-LTER site for transportation to and from sites at the VCR, and we thank Dr. George Luther for help in sampling in Delaware, and Dr. Pat Megonigal for help in sampling in North Carolina. We are indebted to Dave Cleland for help with riboprinting and Paul Krader for help with FAME analysis; in addition we appreciate the assistance of Thuy Pennella-Trang for help with the Bionumerics software, and Mike Peglar for help with some of the sequencing. This work was supported in part by NSF grant DEB-9972099 and a REU supplement from the NSF to L.D.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lydell, C., Dowell, L., Sikaroodi, M. et al. A Population Survey of Members of the Phylum Bacteroidetes Isolated from Salt Marsh Sediments along the East Coast of the United States. Microb Ecol 48, 263–273 (2004). https://doi.org/10.1007/s00248-003-1068-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-003-1068-x