
Abstract
During epidemics, such as the frequent and devastating Ebola virus outbreaks that have historically plagued regions of Africa, serological surveillance efforts are critical for viral containment and the development of effective antiviral therapeutics. Antibody serology can also be used retrospectively for population-level surveillance to provide a more complete estimate of total infections. Ebola surveillance efforts rely on enzyme-linked immunosorbent assays (ELISAs), which restrict testing to laboratories and are not adaptable for use in resource-limited settings. In this manuscript, we describe a paper-based immunoassay capable of detecting anti-Ebola IgG using Ebola virus envelope glycoprotein ectodomain (GP) as the affinity reagent. We evaluated seven monoclonal antibodies (mAbs) against GP—KZ52, 13C6, 4G7, 2G4, c6D8, 13F6, and 4F3—to elucidate the impact of binding affinity and binding epitope on assay performance and, ultimately, result interpretation. We used biolayer interferometry to characterize the binding of each antibody to GP before assessing their performance in our paper-based device. Binding affinity (KD) and on rate (kon) were major factors influencing the sensitivity of the paper-based immunoassay. mAbs with the best KD (3–25 nM) exhibited the lowest limits of detection (ca. μg mL−1), while mAbs with KD > 25 nM were undetectable in our device. Additionally, and most surprisingly, we determined that observed signals in paper devices were directly proportional to kon. These results highlight the importance of ensuring that the quality of recognition reagents is sufficient to support desired assay performance and suggest that the strength of an individual’s immune response can impact the interpretation of assay results.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The frequency and high mortality of Ebola virus outbreaks highlight the pressing need for diagnostic technologies, especially those that can be used for population surveillance. During public health emergencies, such as those induced by the four outbreaks of Ebola virus in the Democratic Republic of the Congo (DRC) that have occurred in the past 4 years (marking outbreaks 8–11 in the DRC alone [1]), accurate and timely surveillance becomes critical [2]. Serological tests, detecting either viral antigens or antibodies raised in response to an infection, can be advantageous when used during or after an epidemic outbreak to aid with viral containment and the development of effective antiviral therapies [2,3,4]. Antibody serology can also be used to survey a population to provide a more complete estimate of total infections, to begin to understand the immune response of survivors if it is not yet known, and to assess the success of virus containment to guide further control measures [2, 5]. When used retrospectively, serological surveillance can gather information about the long-term immune response of survivors, which can inform development of effective and targeted viral therapeutics and vaccines [3]. Additionally, such analyses can be used to better understand trends in exposure among different groups by providing data to aid with the identification of risk factors (e.g., behavioral and demographic) and exposure routes [6].
Currently, there are few methods for Ebola virus serological surveillance that can be used during or after outbreaks, as most of the efforts for the development of diagnostics have been focused on detecting acute infections by RT-PCR or viral antigen assays [7]. Current methods for serological surveillance rely on measuring anti-Ebola IgG using standard enzyme-linked immunosorbent assays (ELISAs) [3, 7,8,9,10]. However, as the complexity of ELISA protocols restricts its use to clinical laboratories, surveillance cannot be performed directly in the field. For widespread surveillance to be feasible for Ebola, or other similar epidemics, the detection method must be suitable for use in limited-resource settings—the areas of Africa that have been impacted by Ebola typically lack access to the laboratory infrastructures required to perform tests. In fact, during the West African Ebola virus epidemic (2014–2016), Ebola surveillance was limited by testing delays caused by over-burdened laboratories, which lacked the capacity and equipment to meet the demand of the outbreak [2, 4]. To that end, the WHO issued a Target Product Profile in 2014 for safe Ebola virus disease diagnostic technologies that can be used at or near the point of use by local staff with minimal training [11]. Paper-based microfluidic technologies are well suited to meet this need, as they offer the potential to be distributed widely, are typically simple to operate, require minimal external instrumentation, and can be designed to provide results at the point of use [12, 13]. Several paper-based device architectures have been designed to support field-deployable immunoassays [14,15,16,17,18,19]. While these approaches include discussions of assay performance, they are primarily focused on the validation of new device design concepts. Components related to device design—involved with controlling reagent storage and fluidics, among other properties—are clearly critical to assay performance. However, they are not the only parameters that impact the intended assay. Specifically, a clear understanding of how the molecular components of a device drive binding (i.e., the interaction between device-loaded affinity reagents and sample analytes) and therefore signal generation would also result in point-of-care tests with improved performance [16].
To study the relationship between affinity reagents and their analytes and ultimately progress towards a point-of-care tool for Ebola surveillance, we developed a paper-based immunoassay that can detect Ebola virus antibodies using the Ebola virus envelope glycoprotein ectodomain (GP), which lacks both the transmembrane and the mucin-like domain, as the affinity reagent. Our device targets anti-Ebola virus IgG, which is typically detectable during the second week of acute illness and known to persist in Ebola virus survivors for many years past exposure [3, 7, 8, 20, 21]. Serum from a convalescent patient will comprise a polyclonal distribution of anti-Ebola IgG with varying affinities, targeting epitopes, and neutralizing capabilities. We therefore screened seven well-characterized monoclonal antibodies (mAbs) against the Ebola virus glycoprotein—KZ52, 13C6, 4G7, 2G4, c6D8, 13F6, and 4F3—to understand how binding affinity and binding epitope influence both assay performance and, ultimately, the interpretation of an individual’s immunity status. We characterized the binding of each antibody to GP using biolayer interferometry (BLI) before assessing their performance in a paper-based immunoassay. We found that both the binding affinity of the antibodies to GP (quantified by KD) and the binding on rate (kon) are the major factors that drive the performance of the paper-based immunoassay. We observed that mAbs with the best KD (ca. nM) had the best limit of detection (ca. μg mL−1), while mAbs with only modest KD (>25 nM) were largely undetected by the paper-based assay. In addition, we observed that the signals generated by modest concentrations of mAb were directly proportional to their kon, which demonstrates an unexpected relationship between a fundamental and measurable property of the analyte/ligand system and overall assay performance. The results presented in this manuscript emphasize the importance of thoroughly characterizing reagent performance to (i) inform assay design and (ii) support desired limits of detection. While we designed these devices with the development of point-of-care serological tools for Ebola in mind, these results may provide insight into key design criteria for rapid assays intended for population surveillance during or after outbreaks of other infectious diseases (e.g., the current COVID-19 pandemic).
Materials and methods
Materials and reagents
We purchased the following mAbs from IBT Bioservices (Rockville, MD): chimeric anti-EBOV GP mAb c6D8, chimeric anti-EBOV GP mAb h13F6, and mouse anti-EBOV GP mAb 4F3. We purchased mouse anti-EBOV mAb clones 2G4 and 4G7 from Sigma-Aldrich. We produced KZ52 in HEK 293F cells from expression plasmids that were a gift from Dennis Burton at Scripps Research Institute. The 13C6 antibody was a gift from Larry Zeitlin at Mapp Biopharmaceutical. We purchased Whatman chromatography paper grade 4 (GE Healthcare Life Sciences) from Amazon. We purchased Biodyne C membrane (0.45-μm pore size) from Pall Corporation. We purchased Flexmount Select DF051521 (permanent adhesive double-faced liner) and Flexmount Select DF021621 (removable/permanent adhesive-double faced liner) from FLEXcon (Spencer, MA). We purchased Fisherbrand chromatography paper (thick) from Fisher Scientific. We purchased Gold-in-a-Box™ conjugation kit (40 nm) from BioAssay Works (Ijamsville, MD). We purchased bovine serum albumin (BSA) and phosphate-buffered saline (PBS, 10×) from Fisher Bioreagents. We purchased Tween 20 and sucrose from Amresco. We purchased casein from EMD Chemicals Inc. We purchased borate-buffered saline (BBS, 5×) from Alfa Aesar. We purchased ACS reagent grade (ASTM type I) water from Ricca Chemical. We purchased Nickel-NTA biosensors and polyclonal Human IgG standards from Pall FortéBio. We purchased single donor human serum from Innovative Research.
Methods
KZ52 synthesis
KZ52 monoclonal antibody was produced via the transfection of plasmids encoding heavy and light chains into FreeStyle 293F cells using polyethylenimine. The day after transfection, cell cultures were supplemented with fresh media and with valproic acid to a final concentration of 2.2 mM. At 6 days post-transfection, supernatant was collected by spin centrifugation and filtration. IgG was isolated using Protein G agarose resin (Thermo Scientific) and subsequently purified via size exchange chromatography using a Superdex 200 Increase 10/300 GL column (GE Healthcare) on an AKTA Pure Protein Purification System (GE Healthcare).
Glycoprotein synthesis and purification
pHLsec-GPΔTM, a gift from Kartik Chandran (Albert Einstein College of Medicine) based in pHL-sec (Addgene plasmid #99845, Edith Yvonne Jones), a mammalian expression plasmid with a secretion signal sequence, was used for expression of GP. pHL-sec-GPΔTM encodes the EBOV GP-Mayinga sequence (UniProt Q05320) with deletions of residues 313–463 of the mucin domain and residues 633–676 of the transmembrane domain. In place of the transmembrane domain are a C-terminal T4 fibritin foldon trimerization peptide (GSGYIPEAPRDGQAYVRKDGEWVLLSTFLGT) and a 6X-His tag. This construct closely resembles a construct used in the first crystallization of unliganded pre-fusion GP (PDB 5JQ3) [22], which we have recreated in previous work [23].
FreeStyle 293F cells (Thermo Fisher) at 2 × 106 cells mL−1 were transfected with 1.5 mg pHLsec-GPΔTM plasmid in a 500-mL culture. At 6 days post-transfection, GP protein was purified from supernatant using PerfectPro Ni-NTA agarose beads (PRIME GmbH). Purified protein was exchanged into phosphate-buffered saline and purified via size exclusion chromatography on a Superdex 200 Increase 10/300 GL column (GE Healthcare). Fractions containing GP were pooled and concentrated to 1 mg mL−1. Aliquots were flash-frozen in liquid nitrogen and stored at −80 °C.
Characterization of binding kinetics by biolayer interferometry (BLI)
We used BLI (K2 Octet, Pall Fortébio) to characterize the binding kinetics between each anti-Ebola mAb and purified GP. Briefly, we used biosensors coated with Nickel-NTA (Pall Fortébio) which chelate to the 6X-His tag on the GP. To determine the proper amount of GP to load onto the sensor, we first performed a loading optimization assay (described in the Supporting Information (ESM)). Once loaded with GP, the sensors are dipped into wells containing a range of concentrations for each mAb. The observed binding rate (kobs) is then measured at each concentration as the mAb associates to the GP. The sensor then moves to a well containing only buffer, which allows the antibody to dissociate from the GP and performs a measurement of the off rate (koff), which is consistent across all concentrations. Baseline steps, where the sensor is dipped into a well containing only buffer, are performed before and after the loading step to assist with data analysis and remove any unbound GP. This process is illustrated in ESM Figure S2. The full protocol is described in more detail in the ESM. The raw association and dissociation curves for each mAb are shown in ESM Figure S3.
Once the assays were completed for each mAb, we plotted kobs against the concentration of mAb. We determined the on rate (kon) by fitting a slope to the plotted data (ESM Figure S4). koff was taken to be the average of the multiple measurements acquired for each assay. Using the ratio of koff to kon, we determined the equilibrium dissociation constant (KD) for each mAb to GP. For negative control experiments using polyclonal human IgG, we followed the same assay protocol as with screening anti-Ebola mAbs (detailed in the ESM). Additionally, we ran a GP-negative control in which we replaced the GP loading well with only buffer (PBS), keeping all other conditions the same. We took the binding magnitude to be the maximum shift (in nm) as measured in the BLI sensorgram.
GP-GC synthesis
We conjugated the GP to 40-nm colloidal gold nanoparticles using the Gold-in-a-Box™ conjugation kit from BioAssay Works, LLC (Ijamsville, MD) to create the glycoprotein-colloidal gold nanoparticle (GP-CG) reporter affinity reagent for our paper assay. We followed the manufacturer’s protocols to first (i) identify the optimal conjugation pH and, subsequently, (ii) synthesize a substantial volume of conjugated colloidal gold nanoparticles to perform our experiments. Briefly, we prepared ten tubes each with 500 μL of colloidal gold nanoparticles at incremental pH spanning 5.4–10.1 using the buffers provided in the kit. Next, we added 14 μL of GP (1 mg mL−1 in 0.5× PBS) to each tube and mixed using a vortexer on a low speed. We allowed the reaction to proceed for 30 min before adding 50 μL of the provided BSA blocking solution to each tube. The theoretical isoelectric point of the GP was calculated to be 5.9 by inputting the full amino acid sequence into an isoelectric point calculator (isoelectric.org). We identified the optimal pH to be 5.4 as the nanoparticles remained in a stable suspension without aggregation; aggregation was visible at higher pH. All batches of GP-CG were produced following the same protocol as described above at pH 5.4 and stored at 4 °C until use. All batches of GP-CG were used within 1 week of synthesis.
Fabrication of paper-based immunoassay device
The wax-printed layers of our three-dimensional paper-based immunoassay device (ESM Figure S5) are designed in Adobe Illustrator and patterned using hydrophobic wax printing (Xerox ColorQube 8580 printer) [24]. We print wax directly on the layers made from cellulose and use a wax-transfer method [25] for the layers made from Biodyne as it is more fragile and therefore susceptible to tearing during direct printing. We melt the wax to form hydrophobic barriers by using a Promo Heat press (PRESS-CS-15) at 280 °F for 45 s to melt the wax through the full thickness of the material. The adhesive layers are designed in Adobe Illustrator and cut using a BOSS LS1630 Laser cutter. We use a double-sided film with both removable and permanent adhesive above the capture layer to facilitate peeling the device upon assay completion. All other layers were assembled using a double-sided permanent adhesive.
Device treatment
We first treated the conjugate layer of the paper device with 2.5 μL of BSA (100 mg mL−1 in 1× PBS), which we allowed to dry at room temperature for 2 min and then at 65 °C for 5 min. We then treated the same zone with 5 μL of 10 OD540 GP-CG, which was diluted from 15 OD540 using a conjugate buffer (200 mg mL−1 sucrose, 20 mg mL−1 BSA, and 0.1% (v/v) Tween 20 in PBS). The determination of the optimal concentration and volume of GP-CG is described in detail in the ESM. The conjugate layer was dried in the same manner as before: first for 2 min at room temperature and then for 5 min at 65 °C. To treat the capture/test readout layer, we first added 5 μL of GP (1 mg mL−1 in 1× PBS) and allowed it to dry at room temperature for 2 min and then at 65 °C for 8 min. Once the GP spot was fully dry, we treated the zones with 6 μL of casein blocking buffer (1% (v/v) casein, 0.05 g mL−1 sucrose, 0.1% (v/v) Tween 20, 1× BBS) and allowed it to dry for 2 min at room temperature and then for 10 min at 65 °C. The scrub layer was treated with 6 μL of casein blocking buffer and allowed to dry at room temperature for 2 min and then at 65 °C for 10 min. We did not treat the sample addition or wash layers.
Device operation
The mAb samples were prepared to desired concentrations (1–40 μg mL−1) by diluting with 1× PBS buffer. Samples containing buffer only were assayed alongside the dilutions of mAb to serve as a negative control for colorimetric background correction. Substantial volume was prepared to allow for the assays to be performed in triplicate at each concentration. To operate the device, we applied 20 μL of sample, containing the desired concentration of mAb, to the sample addition zone on the top layer of the device. Once the sample wicked completely into the device, we added 15 μL of wash buffer (0.05% (v/v) Tween 20 in 1× PBS) and waited for it to wick through before pipetting a second addition (15 μL) of wash buffer to the sample addition zone. When the second wash buffer addition had finished wicking into the device, we peeled apart the device to expose the capture/test readout layer. The devices were immediately scanned using an EPSON Perfection V600 Photo scanner. We performed colorimetric analysis using ImageJ [26]. We employed a similar strategy when the added samples contained polyclonal human IgG or human serum.
Device performance analysis
We quantified color intensity in the green channel of the RGB color space and normalized the results to the values obtained from the negative control devices (buffer only). To calculate the limit of detection (LOD) for each batch of devices, we first determined the average and standard deviations of the intensities of the negative control devices (buffer only) for each batch. Next, we calculated LOD as the magnitude of the response of the blank plus three-times the standard deviation of the blank [27]. We determined the lowest detectable concentration for each mAb to be the lowest concentration that fell above the calculated LOD for each batch of assays.
Results and discussion
Device design
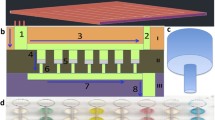
We previously reported a three-dimensional paper-based microfluidic device architecture to perform traditional sandwich immunoassays [14]. We later adapted this architecture to perform a double-antigen sandwich immunoassay (i.e., detecting antibodies in a sample only when bound by two antigens), which we demonstrated to have superior analytical performance when compared to other indirect immunoassay formats [28]. Therefore, we employ the double-antigen sandwich immunoassay format here. Briefly, the device is fabricated using seven layers with unique roles: (i) sample addition, (ii) conjugate storage, (iii) incubation, (iv) scrub, (v) capture/test readout, (vi) wash, and (vii) blot (Fig. 1a). When the sample, containing anti-Ebola virus mAb, is added to the sample addition layer, it wicks vertically to the subsequent conjugate storage layer, where it rehydrates the glycoprotein-colloidal gold nanoparticle conjugate (GP-CG) that is stored there. Next, the sample wicks to the incubation layer, where it travels laterally and allows the antibodies to mix with and bind to the GP-CG to form the desired, partial immunocomplex (mAb-GP-CG). The sample subsequently wicks vertically through the scrub layer, which filters any aggregated GP-CG particles from the sample to reduce non-specific capture of the gold conjugate [28]. The partial immunocomplex is then transported to the capture/test readout layer, which is treated with GP to capture mAb-GP-CG and form the full detection complex (Fig. 1b). The mAb can thus be detected colorimetrically from the visible signal produced by the captured colloidal gold. The remaining fluid is wicked vertically to the wash layer, which directs excess fluid to the blot layer below. The materials used for each layer and the treatments for the conjugate storage, scrub, and capture/test readout layers are described further in detail in the ESM.

Design of the paper device for conducting an Ebola immunoassay. a Device schematic showing each layer and its function. The added sample contains anti-Ebola virus mAbs, followed by two additions of wash buffer. The sample wicks vertically to the conjugate storage layer, where it rehydrates the glycoprotein-colloidal gold (GP-CG) reporter reagent that is stored there. The mAbs incubate with the GP-CG, forming the desired partial immunocomplex. The scrub layer serves to remove any colloidal gold aggregates. The capture/readout layer is treated with the recombinant Ebola glycoprotein construct, GP, to capture the mAb-GP-CG and form the full immunocomplex. The wash and blot layers serve to wick excess fluid. b Double-antigen sandwich immunoassay format. A colorimetric signal is produced by the colloidal gold on the capture/readout layer, corresponding to the amount of antibody captured
Selection of anti-Ebola monoclonal antibodies
We chose to characterize the performance of our paper-based assay using seven monoclonal antibodies that have been well-studied for their reactivity to Ebola GP. KZ52 is a neutralizing antibody that was isolated from a human survivor in 1999 [29] and is known to bind to the GP base [30,31,32]. 13C6 (murine/human chimera) is a component of the MB-003 and Zmapp therapeutic cocktails and binds to the glycan cap of GP [32, 33]. The mAbs 4G7 and 2G4 (both murine) are components of the ZMAb and Zmapp therapeutic cocktails and, like KZ52, bind to the GP base [32]. The mAbs c6D8 (murine/human chimera) and 13F6 (murine/human chimera) are components of the MB-003 cocktail and bind to the mucin-like domain of GP, which is not present in our GP construct [32, 33]. Lastly, 4F3 is a non-neutralizing murine antibody to GP, and its epitope lies within the mucin domain [31]. The epitopes for each of the seven mAbs are highlighted in Fig. 2.

Ebola GP structure with highlighted antibody-binding epitopes. a Protein construct of Ebola GP, with GP1 in cyan and GP2 in dark blue. Disulfide bonds are shown linking GP1 and GP2, and the GP1 glycan cap and mucin-like domain are highlighted. Of note, the C-terminal transmembrane domain has been removed. b Crystal structure of Ebola GP trimer with mucin-like domains modeled in gray (PDB: 5JQ3) [22]. Binding epitopes for monoclonal antibodies 13F6, 4F3, and c6D8 (light blue); 13C6 (yellow); and 2G4, 4G7, and KZ52 (overlapping, red) are highlighted
By selecting a variety of mAbs that bind to distinct domains of Ebola GP, we expected to measure a wide range of binding affinities to help us better understand the role of immunoreactivity on the performance of paper-based immunoassays. We anticipated that the three antibodies that have been characterized to primarily recognize the mucin-like domain (c6D8, 13F6, and 4F3) could serve as our negative controls as they would likely exhibit poor binding to our GP construct, which lacks the mucin-like domain. Additionally, using a selection of mAbs that bind to both distinct epitopes in three separate domains of Ebola GP (i.e., GP base, glycan cap, and mucin-like domain) and also overlapping epitopes (i.e., on the GP base for KZ52, 2G4, and 4G7), we hoped to learn more about the role of epitope on assay performance.
Binding parameters for mAbs to GP
We determined the binding kinetic parameters by BLI using Ni-NTA biosensors loaded with C-terminal 6X-His-tagged GP. Table 1 details the compiled binding kinetic parameters for each mAb to GP, where calculated errors in KD are determined by propagating errors associated with measured off rates (koff) and calculated on rates (kon). The raw binding curves for each mAb are shown in ESM Figure S3. In summary, KZ52 exhibited the strongest binding to GP with 3.8 nM KD and demonstrated the fastest kon and the slowest koff of the seven mAbs screened. 13C6 exhibited the second-best binding with a KD of 10.9 nM. mAbs 4G7 and 2G4 demonstrated nearly identical binding affinities at 24.0 nM and 24.3 nM, respectively, where they were also characterized by similar kon and koff. 4F3 and c6D8 exhibited very similar binding affinities at 53.2 nM and 59.9 nM, respectively, but had twofold differences in koff. Lastly, 13F6 exhibited the worst binding to GP at 205.0 nM.
Out of the seven mAbs we selected, KZ52 has been shown throughout the literature to demonstrate high affinity to GP, with reported KD around 2 nM [32]. Therefore, our result of 3.8 nM binding is reasonable and consistent with previous reports. The KD we obtained for 13C6 of 10.9 nM also falls within the range of published KD values, which span from 0.4 to 47 nM for several variants of GP [32, 34, 35]. Published apparent affinities for 2G4 and 4G7 are stronger than the values we measured using BLI experiments: ~3 nM for 2G4 and ~1 nM for 4G7 [32]. However, these values of KD were determined by ELISA, which is known to provide only a relative estimate of binding. Davidson et al. demonstrated that mAbs KZ52, 13C6, 2G4, and 4G7 showed strong apparent affinities to the GP variant most like our own, the glycoprotein without the mucin-like domain (GP∆Mucin), while mAbs c6D8 and 13F6 had binding affinities that were undetectable with the same construct [32]. Our own characterizations by BLI show that mAbs c6D8 and 13F6 exhibit detectable, yet weak binding to our GP construct, with measured KDs of 59.9 nM and 205.0 nM, respectively. To our knowledge, there are no published KD values for 4F3 to GP against which to compare our measurement (53.2 nM). The results of binding assays using mAbs 4F3, c6D8, and 13F6 were unexpected based on our understanding of their characterized epitopes, which reside in the mucin-like domain and is not present in our GP construct. However, as seen in the individual BLI sensorgrams for these mAbs (ESM Figure S3), this weak binding is consistent with a specific interaction. These data suggest these antibodies may interact, albeit weakly, with GP outside of the mucin-like domain.
Performance of paper-based immunoassay for all mAbs
We characterized the analytical performance of the capture-sandwich format of our paper-based immunoassay device for all seven Ebola mAbs. Due to the scales at which assay reagents (recombinant GP and conjugated GP-CG) could be prepared and the amounts that were required to conduct experiments, we evaluated assays in two separate batches: (i) KZ52, 13C6, c6D8, 4F3, and 13F6 and (ii) KZ52, 2G4, and 4G7. KZ52 was evaluated in both device batches in order to provide a validated comparator. We assessed detection with mAbs diluted to the following concentrations: 1, 5, 10, 20, and 40 μg mL−1. We selected this range based on published concentrations in neutralization studies for Ebola mAbs [29, 36, 37].
Paper-based immunoassays are completed in ~25 min, following addition of sample (20 μL) and two aliquots of wash buffer (2× 15 μL). When the wash buffer had completely wicked into the sample layer, we peeled the device to arrest flow and expose the test zone. We scanned test zones and analyzed them for color intensity using ImageJ. Figure 3a shows representative scans for three of the five mAbs from the first batch: KZ52, 13C6, and 13F6, demonstrating strong, moderate, and no signal, respectively. The quantitative results for each antibody from the first batch are shown graphically in Fig. 3b. KZ52 provided the strongest signal across all concentration ranges compared to the other mAbs and was detectable even at the lowest screened concentration of 1 μg mL−1. mAb 13C6 was also detected in the paper immunoassay. However, the signals for 13C6 were weaker than those of KZ52 across all concentrations. While the signal at 1 μg mL−1 fell below the limit of detection (LOD) of the assay, calculated as the magnitude of the response of the blank plus three-times the standard deviation of the blank, signals were detectable at the remaining concentrations across the range from 5 to 40 μg mL−1. The other mAbs, 4F3, c6D8, and 13F6, were not detected in our device format, with no discernable signal across the selected concentration range (i.e., all signals fell below the LOD). Figure 3c shows representative scans from the second batch of mAb assays for KZ52, 4G7, and 2G4, respectively. Results from ImageJ analysis for calibration curves from each antibody are shown graphically in Fig. 3d. The signal produced by KZ52 in this batch was weaker than that from the first batch of testing, which we can attribute to lot-to-lot variation of the GP-CG conjugate. The LOD was also slightly higher in this batch of testing, so the lowest detectable concentration for KZ52 was 5 μg mL−1. In comparison to KZ52, 4G7 showed weaker signal across all concentrations, but was detectable down to 20 μg mL−1 in this format. mAb 2G4 had the worst performance of the second batch, exhibiting very little to no signal across most of the concentration range, but showed a detectable signal at 40 μg mL−1.

Paper-based immunoassay results for all mAbs. a Representative scans of test zones from 0 to 40 μg mL−1 for mAbs KZ52, 13C6, and 13F6 demonstrating strong, moderate, and no signal, respectively. b Quantitative results from ImageJ analysis for each mAb in the first batch of assays (KZ52, 13C6, 4F3, c6D8, 13F6), n = 3. c Representative scans of test zones from 0 to 40 μg mL−1 for mAbs KZ52, 4G7, and 2G4. d Quantitative results from ImageJ analysis in the second batch of assays (KZ52, 2G4, and 4G7), n = 3. Error bars indicate standard deviation
The results from the paper-based immunoassay are summarized in Table 2, where we also provide the corresponding binding parameters (kon, koff, KD) for each of the seven mAbs to facilitate comparisons. Briefly, KZ52, with the lowest KD of 3.8 nM, exhibited the lowest detectable concentration (1 μg mL−1), followed by 13C6, with a KD of 10.9 nM and detectable signal at a concentration of 5 μg mL−1. Both 4G7 and 2G4 showed the weakest, yet measurable signals (detectable at 20 and 40 μg mL−1), with KDs of 24.0 nM and 24.3 nM, respectively. The other mAbs (4F3, c6D8, and 13F6), with binding affinities greater than 50 nM, were undetectable by our device across the entire range of concentrations tested.
To further elucidate the trends between the kinetic parameters and the results of the paper immunoassay, we calculated the Pearson correlation coefficients between each parameter and (i) the lowest detectable concentration (μg mL−1) and (ii) the observed signal intensity at 20 μg mL−1 (a.u.). These results are shown in Table 3. The results from the paper-based immunoassay mirror the trend in the equilibrium dissociation constant of each mAb to our GP construct: a lower KD has a strong positive correlation to a lower detectable signal (r = 0.88) and a modest inverse correlation to observed signal (r = −0.56). However, a more striking result from these analyses is the very strong positive association between kon and the observed signal at 20 μg mL−1 (r = 0.97), which suggests a direct, linear relationship between them (ESM Figure S6). The on rate also shows a strong inverse association with the lowest detectable signal (r = −0.87). Like with KD, there is a cutoff in magnitude of kon at which mAbs are no longer detectable in our device: around 2.0 × 10−5 nM−1 s−1. The off rate shows a relatively strong inverse correlation with the observed signal at 20 μg mL−1 (r = −0.76), and a modest positive correlation to a lower detectable signal (r = 0.60). These results indicate that both kon and koff, when taken individually, are effective predictors of observed signal intensity, and kon, specifically, is a very strong predictor of immunoassay performance.
The relationship between KD and assay performance validates what is already known conceptually to most immunoassay developers. Even though KD can be obtained from the ratio of off and on rates, it is fundamentally an equilibrium constant: the ratio of the concentrations of free analyte and ligand (numerator) to the concentration of bound complex (denominator) when the binding interaction has reached equilibrium. An analyte with lower KD would indicate a higher concentration of bound complex—and more signal generated—than one with a higher KD. Thus, assay developers often choose reagents with the lowest KD. In our paper device, however, equilibrium between analyte (mAb) and affinity reagent (GP-CG or spotted GP) is not reached because ideal conditions for equilibrium (e.g., lengthy reaction times with unchanging reagent concentrations in a static system) cannot feasibly be replicated in our paper device. Specifically: (i) the assay duration is short (~25 min); (ii) the partial immunocomplex is formed under flow due to wicking by capillary action and likely with incompletely rehydrated GP-CG, which results in variances in effective reagent concentration; and (iii) the final completed immunocomplex (on the test zone) is also formed under flow due to wicking. Even without reaching equilibrium, we show that KD is a reasonable assay parameter to optimize around. However, we hypothesize that kon serves as the best predictor of assay performance in the paper-based device because it is related to the amount of signal-generating complex that can be formed while the assay is being conducted. With knowledge of this relationship, changes to the design of the paper device (e.g., increasing channel length [14], introducing structures that control flow and incubation times [38, 39]) to increase assay time could, theoretically, improve signal generation by promoting an increase in complex formation. However, such changes may come at the cost of assay duration, manufacturability, and potentially usability [40]. To the best of our knowledge, the direct relationship between on rate and signal production has not yet been demonstrated and offers new opportunities for assay design and optimization for a variety of immunoassay devices that rely on wicking (i.e., paper devices or lateral flow devices).
Contribution of molecular recognition to assay performance
We observed an unexpected difference in the performance of paper-based immunoassays for mAbs 4G7 and 2G4, which are characterized by almost identical KD (24.0 nM vs. 24.3 nM, respectively) and kon (2.6 × 10−5 nM−1·s−1 vs. 2.3 × 10−5 nM−1·s−1, respectively). In paper-based immunoassays, 4G7 was detectable at a lower concentration than mAb 2G4 (20 μg mL−1 vs. 40 μg mL−1) and produced a much more intense signal at 20 μg mL−1 (Table 2). These results suggest that the difference in assay performance is not due to inherent affinity between mAb and GP, but perhaps to the assay format itself controlling the molecular recognition events required to form the complete and detectable immunocomplex between GP-CG, mAb, and capture-zone adsorbed GP.
It is known that 4G7 and 2G4 bind to similar regions of the base of GP but using unique epitopes [32]. Davidson et al. determined that mAb 2G4 uses critical residues C511, N550, G553, and C556 while mAb 4G7 instead uses critical residue D552 to bind to the GP base. Qiu et al. demonstrated in a Western blot that 4G7 recognized GP but 2G4 did not, suggesting that the epitope for 2G4 is conformational [41]. BLI experiments utilized scaffold engineering where the GP is first immobilized to the Ni-NTA biosensor by a C-terminal 6X-His tag, resulting in a consistent orientation of the GP. This orientation on the surface of a planar sensor tip presumably allows sufficient access of both mAbs 2G4 and 4G7 to their epitopes on the GP base, resulting in very similar KD measurements. However, to prepare these paper-based immunoassays, we rely on physisorption of GP to both create the GP-CG reporter and coat the capture membrane. Since physisorption is largely an unpredictable process with no directed control over molecular orientation on either the spherical gold particle or fibers of the Biodyne membrane (a Nylon-based material), we cannot assume that GP is presented to solvent in a manner that maximizes binding potential or facilitates equivalent access to all antigenic epitopes. Additionally, it is possible that adsorption distorts the conformation of GP epitopes to an extent, which is a recognized potential consequence of protein adsorption to solid surfaces [42, 43] and, specifically, nanoparticle surfaces [44, 45]. We therefore speculate that a subtle difference in the availability and conformation of GP epitopes—on GP-CG, the capture membrane, or both—could be a cause of the observed difference in performance of paper-based immunoassays for mAbs with similar KD, as we saw with mAbs 4G7 and 2G4. Further work would be necessary to fully characterize and understand the relationship between epitope presentation (e.g., via further reagent engineering or scaffolding [46]) and assay performance. Such results would not only offer insight into how to design paper-based immunoassays but also more broadly into how immunoreactivity to antigens manifests in devices designed for serological surveillance.
Evaluation of assay specificity with complex sample matrices
To confirm the specificity of the assay for anti-Ebola mAbs, we first used BLI to determine whether any non-specific binding occurs between off-target mAbs and the GP. We prepared dilutions of polyclonal human IgG at the same concentrations tested in the paper immunoassay (1, 5, 10, 20, and 40 μg mL−1) to serve as true negative controls. BLI experiments were performed as detailed in the ESM. By analyzing the observed binding rate (kobs) across the range of concentrations, it became evident that no detectable, specific binding occurred between the polyclonal human IgG and the GP (ESM Figure S7). Although the sensorgrams showed binding, this was confirmed to be non-specific binding to the Ni-NTA biosensor and not to the GP (ESM Figures S7–S9): (i) the slope of kobs vs. concentration of polyclonal IgG was slightly negative; (ii) the magnitude of binding (expressed in nm) was directly proportional to concentration; and (iii) the magnitude of binding was equal for IgG exposed to tips both functionalized and unfunctionalized with GP. Therefore, the binding events observed for the seven anti-Ebola mAbs were confirmed to be specific even when weakly binding mAbs had only putative epitopes on our GP construct (e.g., clone 13F6).
To assess assay specificity in the paper-based device and to gauge assay performance with sample matrices more complex than buffer, we tested the devices using (i) negative samples composed of 20 μg mL−1 polyclonal human IgG and undiluted human serum and (ii) contrived “positive” samples of 20 μg mL−1 polyclonal human IgG and undiluted human serum spiked with 20 μg mL−1 KZ52. Samples in PBS only were used as comparator controls. All assays were performed in triplicate and the results from these experiments are shown in Fig. 4. The time to assay completion was longer for the serum samples (~45 min) than those for more simple matrices comprising only polyclonal human IgG or PBS (~25 min), which is expected due to its increased viscosity and protein content. We observed that samples containing a background of polyclonal human IgG produced almost identical intensities for both the positive and negative specimens to those of PBS. The lack of signal produced by the negative control further validated the results from BLI, confirming the high assay specificity of our GP construct for anti-Ebola mAbs. Additionally, the signal produced in the positive specimen demonstrated that recovery of KZ52 binding is retained in the presence of off-target IgG. Recovery of KZ52 binding was equally successful in contrived samples prepared from healthy human serum. Assays conducted using human serum had a slightly higher background in the negative control (i.e., serum only) compared to the negative controls for other conditions (i.e., PBS or polyclonal human IgG lacking KZ52). We expect this background could be reduced in future assays by modifying blocking or washing conditions, which were optimized here only for samples in PBS. Positive samples containing spiked KZ52 experienced almost equal increases in signal in all matrices: 60.1 ± 3.5 a.u. for serum, 62.0 ± 1.4 a.u. for polyclonal human IgG, and 63.0 ± 1.2 a.u. for PBS. These results not only confirmed assay specificity for anti-Ebola mAbs but also demonstrated the successful detection of target, antigen-specific IgG in an appropriate sample matrix, human serum.

Sample matrix comparison in paper-based immunoassay devices. Quantitative ImageJ results for contrived positive samples (spiked with 20 μg mL−1 KZ52) and negative controls (containing no KZ52) in PBS, polyclonal human IgG (20 μg mL−1), and undiluted human serum. Representative scans of test zones for each condition are shown above the corresponding bar graph. Error bars indicate standard deviation (n = 3)
Conclusions
In this work, we employ a double-antigen sandwich immunoassay for the colorimetric detection of anti-Ebola virus IgG in a paper-based device using recombinant Ebola glycoprotein as both capture and reporter affinity reagents. The results presented here lay the groundwork for the development of a point-of-care immunoassay to aid with Ebola surveillance either during an outbreak, to provide timely and critical information for viral containment, as well as retrospectively to gather immune response data at the population level. While further development and validation of this assay with more complex biological samples (e.g., convalescent plasma from Ebola survivors) are required to bring this specific device closer to its intended use, the observations we describe here have broader implications in the creation and interpretation of the performance of point-of-care serological tests—namely, that assay results can be related to binding affinity (and potentially epitope accessibility) of affinity reagents, and are not solely dependent on the concentration of antibodies in the sample. Conceptually, this conclusion is evident to immunoassay developers. However, to our knowledge, this is the first study that compares the binding of a series of well-characterized mAbs over a range of KD directly to their performance in a device designed to conduct point-of-care immunoassays. Additionally, the more surprising results of this work are the derived direct relationship between kon and observed signal, and the potential implications for optimizing assay performance when using binding rates as selection criteria for affinity reagents. These results inform guidance for the design of effective point-of-care assays by demonstrating the practical importance of characterizing binding—via rates, overall affinity, and epitope accessibility—in addition to the potential need to evaluate conjugation strategies beyond physisorption when developing recognition reagents.
Critically, these observations also have the potential to greatly impact the interpretation of surveillance efforts. Specifically, false negatives for tests of this type could occur if an individual had poor immunoreactivity (i.e., low antibody titer or persistent clones with low affinities). False negatives in population surveillance are undesirable when the goal of surveillance is to gain an accurate estimate of total infections. However, in some cases, a false negative could be an acceptable diagnostic outcome: those individuals testing negative could lack adequate protection against reinfection and could be at risk for subsequent exposures. These results are not limited in application to Ebola virus surveillance—adequate surveillance could help determine policies around social isolation, self-quarantine, and return-to-work practices for the current COVID-19 pandemic.
References
Years of Ebola virus disease outbreaks - CDC, 2020. https://www.cdc.gov/vhf/ebola/history/chronology.html. Accessed December 16, 2020.
McNamara LA, Schafer IJ, Nolen LD, Gorina Y, Redd JT, Lo T, et al. Ebola surveillance - Guinea, Liberia, and Sierra Leone. MMWR Suppl. 2016;65:35–43. https://doi.org/10.15585/mmwr.su6503a6.
Rimoin AW, Lu K, Bramble MS, Steffen I, Doshi RH, Hoff NA, et al. Ebola virus neutralizing antibodies detectable in survivors of the Yambuku, Zaire outbreak 40 years after infection. J Infect Dis. 2018;217:223–31. https://doi.org/10.1093/infdis/jix584.
Houlihan CF, Youkee D, Brown CS. Novel surveillance methods for the control of Ebola virus disease. Int Health. 2017;9:139–41. https://doi.org/10.1093/inthealth/ihx010.
CDC, COVID-19 serology surveillance strategy. n.d.. https://www.cdc.gov/coronavirus/2019-ncov/covid-data/serology-surveillance/index.html. Accessed November 5, 2020.
Dietz PM, Jambai A, Paweska JT, Yoti Z, Ksaizek TG. Epidemiology and risk factors for Ebola virus disease in Sierra Leone - 23 May 2014 to 31 January 2015. Clin Infect Dis. 2015;61:1648–54. https://doi.org/10.1093/cid/civ568.
Broadhurst MJ, Brooks TJG, Pollock NR. Diagnosis of Ebola virus disease: past, present, and future. Clin Microbiol Rev. 2016;29:773–93. https://doi.org/10.1128/CMR.00003-16.
Ksiazek TG, West CP, Rollin PE, Jahrling PB, Peters CJ. ELISA for the detection of antibodies to Ebola viruses. J Infect Dis. 1999;179:S192–8. https://doi.org/10.1086/514313.
Busico KM, Marshall KL, Ksiazek TG, Roels TH, Fleerackers Y, Feldmann H, et al. Prevalence of IgG antibodies to Ebola virus in individuals during an Ebola outbreak, Democratic Republic of the Congo, 1995. J Infect Dis. 1999;179:S102–7. https://doi.org/10.1086/514309.
Clark DV, Kibuuka H, Millard M, Wakabi S, Lukwago L, Taylor A, et al. Long-term sequelae after Ebola virus disease in Bundibugyo, Uganda: a retrospective cohort study. Lancet Infect Dis. 2015;15:905–12. https://doi.org/10.1016/S1473-3099(15)70152-0.
World Health Organization (WHO). Target product profile for Zaïre ebolavirus rapid, simple test to be used in the control of the Ebola outbreak in West Africa. 2013.
Martinez AW, Phillips ST, Whitesides GM, Carrilho E. Diagnostics for the developing world: microfluidic paper-based analytical devices. Anal Chem. 2010;82:3–10. https://doi.org/10.1021/ac9013989.
Fernandes SC, Walz JA, Wilson DJ, Brooks JC, Mace CR. Beyond wicking: expanding the role of patterned paper as the foundation for an analytical platform. Anal Chem. 2017;89:5654–64. https://doi.org/10.1021/acs.analchem.6b03860.
Schonhorn JE, Fernandes SC, Rajaratnam A, Deraney RN, Rolland JP, Mace CR. A device architecture for three-dimensional, patterned paper immunoassays. Lab Chip. 2014;14:4653–8. https://doi.org/10.1039/c4lc00876f.
Yee EH, Lathwal S, Shah PP, Sikes HD. Detection of biomarkers of periodontal disease in human saliva using stabilized, vertical flow immunoassays. ACS Sensors. 2017;2:1589–93. https://doi.org/10.1021/acssensors.7b00745.
Sung KJ, Al Maalouf YJ, Johns QR, Miller EA, Sikes HD. Functional comparison of paper-based immunoassays based on antibodies and engineered binding proteins. Analyst. 2020;145:2515–9. https://doi.org/10.1039/d0an00299b.
Phillips EA, Young AK, Albarran N, Butler J, Lujan K, Hamad-Schifferli K, et al. Ampli: a construction set for paperfluidic systems. Adv Healthc Mater. 2018;7:1–8. https://doi.org/10.1002/adhm.201800104.
Pollok NE, Rabin C, Walgama CT, Smith L, Richards I, Crooks RM. Electrochemical detection of NT-proBNP using a metalloimmunoassay on a paper electrode platform. ACS Sensors. 2020;5:853–60. https://doi.org/10.1021/acssensors.0c00167.
Cunningham JC, Scida K, Kogan MR, Wang B, Ellington AD, Crooks RM. Paper diagnostic device for quantitative electrochemical detection of ricin at picomolar levels. Lab Chip. 2015;15:3707–15. https://doi.org/10.1039/c5lc00731c.
Ksiazek TG, Rollin PE, Williams AJ, Bressler DS, Martin ML, Swanepoel R, et al. Clinical virology of Ebola hemorrhagic fever (EHF): virus, virus antigen, and IgG and IgM antibody findings among EHF patients in Kikwit, Democratic Republic of the Congo, 1995. J Infect Dis. 1999;179:S177–87. https://doi.org/10.1086/514321.
Wauquier N, Becquart P, Gasquet C, Leroy EM. Immunoglobulin G in Ebola outbreak survivors, Gabon. Emerg Infect Dis. 2009;15:1126–37. https://doi.org/10.1086/514290.
Zhao Y, Ren J, Harlos K, Jones DM, Zeltina A, Bowden TA, et al. Toremifene interacts with and destabilizes the Ebola virus glycoprotein. Nature. 2016;535:169–72. https://doi.org/10.1038/nature18615.Toremifene.
Durham ND, Howard AR, Govindan R, Senjobe F, Fels JM, Diehl WE, et al. Real-time analysis of individual Ebola virus glycoproteins reveals pre-fusion, entry-relevant conformational dynamics. Viruses. 2020;12:1–15.
Carrilho E, Martinez AW, Whitesides GM. Understanding wax printing: a simple micropatterning process for paper-based microfluidics. Anal Chem. 2009;81:7091–5. https://doi.org/10.1021/ac901071p.
Lu Y, Lin B, Qin J. Patterned paper as a low-cost, flexible substrate for rapid prototyping of PDMS microdevices via liquid molding. Anal Chem. 2011;83:1830–5. https://doi.org/10.1021/ac102577n.
Schneider CA, Rasband WS, Eliceiri KW. NIH image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. https://doi.org/10.1038/nmeth.2089.
Shrivastava A, Gupta V. Methods for the determination of limit of detection and limit of quantitation of the analytical methods. Chron Young Sci. 2011;2:21. https://doi.org/10.4103/2229-5186.79345.
Fernandes SC, Logounov GS, Munro JB, Mace CR. Comparison of three indirect immunoassay formats on a common paper-based microfluidic device architecture. Anal Methods. 2016;8:5204–11. https://doi.org/10.1039/c6ay01558a.
Maruyama T, Rodriguez LL, Jahrling PB, Sanchez A, Khan AS, Nichol ST, et al. Ebola virus can be effectively neutralized by antibody produced in natural human infection. J Virol. 1999;73:6024–30. https://doi.org/10.1128/jvi.73.7.6024-6030.1999.
Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature. 2008;454:177–82. https://doi.org/10.1038/nature07082.
Panina AA, Dementieva IG, Aliev TK, Toporova VA, Balabashin DS, Bokov MN, et al. Recombinant antibodies to the Ebola virus glycoprotein. Acta Nat. 2017;9:84–91. https://doi.org/10.32607/20758251-2017-9-4-84-91.
Davidson E, Bryan C, Fong RH, Barnes T, Pfaff JM, Mabila M, et al. Mechanism of binding to Ebola virus glycoprotein by the ZMapp, ZMAb, and MB-003 cocktail antibodies. J Virol. 2015;89:10982–92. https://doi.org/10.1128/jvi.01490-15.
Murin CD, Fusco ML, Bornholdt ZA, Qiu X, Olinger GG, Zeitlin L, et al. Structures of protective antibodies reveal sites of vulnerability on ebola virus. Proc Natl Acad Sci U S A. 2014;111:17182–7. https://doi.org/10.1073/pnas.1414164111.
Misasi J, Gilman MSA, Kanekiyo M, Gui M, Cagigi A, Mulangu S, et al. Structural and molecular basis for Ebola virus neutralization by protective human antibodies. Science. 2016;351:1343–6. https://doi.org/10.1126/science.aad6117.
Rijal P, Elias SC, Machado SR, Xiao J, Schimanski L, O’Dowd V, et al. Therapeutic monoclonal antibodies for Ebola virus infection derived from vaccinated humans. Cell Rep. 2019;27:172–186.e7. https://doi.org/10.1016/j.celrep.2019.03.020.
Liu Q, Fan C, Li Q, Zhou S, Huang W, Wang L, et al. Antibody-dependent-cellular-cytotoxicity-inducing antibodies significantly affect the post-exposure treatment of Ebola virus infection. Sci Rep. 2017;7:1–11. https://doi.org/10.1038/srep45552.
McMullan LK, Flint M, Chakrabarti A, Guerrero L, Lo MK, Porter D, et al. Characterisation of infectious Ebola virus from the ongoing outbreak to guide response activities in the Democratic Republic of the Congo: a phylogenetic and in vitro analysis. Lancet Infect Dis. 2019;19:1023–32. https://doi.org/10.1016/S1473-3099(19)30291-9.
Martinez AW, Phillips ST, Nie Z, Cheng CM, Carrilho E, Wiley BJ, et al. Programmable diagnostic devices made from paper and tape. Lab Chip. 2010;10:2499–504. https://doi.org/10.1039/c0lc00021c.
Lutz B, Liang T, Fu E, Ramachandran S, Kauffman P, Yager P. Dissolvable fluidic time delays for programming multi-step assays in instrument-free paper diagnostics. Lab Chip. 2013;13:2840–7. https://doi.org/10.1039/c3lc50178g.
Murray LP, Mace CR. Usability as a guiding principle for the design of paper-based , point- of-care devices: a review. Anal Chim Acta. 2020;1140:236–49. https://doi.org/10.1016/j.aca.2020.09.063.
Qiu X, Alimonti JB, Melito PL, Fernando L, Ströher U, Jones SM. Characterization of Zaire ebolavirus glycoprotein-specific monoclonal antibodies. Clin Immunol. 2011;141:218–27. https://doi.org/10.1016/j.clim.2011.08.008.
Hlady V, Buijs J. Protein adsorption on solid surfaces. Curr Opin Biotechnol. 1996;7:72–7.
Thyparambil AA, Wei Y, Latour RA. Experimental characterization of adsorbed protein orientation, conformation, and bioactivity. Biointerphases. 2015;10:019002. https://doi.org/10.1116/1.4906485.
Wangoo N, Suri CR, Shekhawat G. Interaction of gold nanoparticles with protein: a spectroscopic study to monitor protein conformational changes. Appl Phys Lett. 2008;92:1–4. https://doi.org/10.1063/1.2902302.
Roach P, Farrar D, Perry CC. Surface tailoring for controlled protein adsorption: effect of topography at the nanometer scale and chemistry. J Am Chem Soc. 2006;128:3939–45. https://doi.org/10.1021/ja056278e.
Cavazos-Elizondo D, Sung K-J, Vasikaran S, Aguirre-Soto A, Sikes HD. Functional comparison of bioactive cellulose materials incorporating engineered binding proteins. ACS Appl Bio Mater. 2021. https://doi.org/10.1021/acsabm.0c01474.
Acknowledgements
The project described was supported by the Tufts University Office of the Vice Provost for Research (OVPR) Tufts Collaborates Seed Grant Program, NIH grant DP2 AI124384, National Science Foundation grant CBET-1846846, and a generous gift from James Kanagy.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
ABC Highlights: authored by Rising Stars and Top Experts.
Supplementary information
ESM 1
(PDF 735 kb)
Rights and permissions
About this article

Cite this article
Murray, L.P., Govindan, R., Mora, A.C. et al. Antibody affinity as a driver of signal generation in a paper-based immunoassay for Ebola virus surveillance. Anal Bioanal Chem 413, 3695–3706 (2021). https://doi.org/10.1007/s00216-021-03317-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-021-03317-4