
Abstract
The diffusion coefficient of glucose in different media is an important parameter in life sciences, as well as in biotechnology and microbiology. In this work a simple, fast method is proposed that is based on the electrochemical time of flight principle. In most of the earlier time of flight experiments performed, a constant flight distance was applied. In the present work a scanning electrochemical microscope (SECM) was applied as a measuring tool. With use of the SECM, the flying distance could be changed with high precision, making measurements with several flight distances more accurate and reliable values could be obtained for solutions as well as for gels. The conventional voltammetric methods are not applicable for glucose detection. In our work electrocatalytic copper oxide coated copper microelectrodes and micro-sized amperometric enzyme sensors were used as detectors, while microdroplet-ejecting pneumatically driven micropipettes were used as a source.

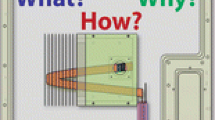
Experimental set up for SCEM-TOF diffusion coefficient measurements
Similar content being viewed by others

Introduction
The transport rate of glucose, an important energy substrate in biochemical processes, can influence different biochemical or physiological processes involving glucose uptake. A high number of measurement devices [1] and methods have been developed for measurement of glucose transport. Different mediated active transport mechanisms [2, 3] are involved in living plant or animal tissues and cell suspensions and control the partitioning or uptake of glucose. However, the importance of passive transport, diffusion, driven by chemical potential difference in quiescent media is obvious.
For measurements of diffusion coefficients in different membranes or gels, often two-compartment diffusion cells are used. In those studies steady state, or quasi steady state, and time of lag principles can be used. Measurements with diffusion cells of different design are usually slow and complicated. The diffusion medium has to be isolated and introduced into, or prepared in situ in, the cell. Its thickness has to be known and kept constant. The concentration of diffusing species has to be followed in time.
Zhang and Furusaki [4] compared the diffusion cell techniques for glucose. They concluded that in case of the time of lag method, the intercept of the concentration-time plot can give erroneously low diffusion coefficient values.
As is well known, the diffusion coefficient of electroactive species is one of the basic parameters controlling the current in voltammetric experiments. Therefore, several methods based on electrochemical measurements, such as polarography, chronoamperometry, and DC voltammetry, have been developed and used for the determination of the diffusion coefficient of electroactive species. The electrode processes are usually complex reactions of several consecutive steps; therefore, voltammetric measurements give reliable diffusion coefficient values only if diffusion is the rate-determining step.
When application of electrochemical diffusion coefficient measurement methods is considered for glucose, difficulties can be foreseen. On one hand, the glucose does not show electric activity in the potential window available in the case of carbon electrodes. On the other hand, gold electrodes become deactivated, fouled by the product of the electrochemical reaction.
Copper or nickel electrodes can be used for voltammetric determination of glucose and other saccharides in basic media. Owing to the electrocatalytic nature of the metal oxide films coating the surface of these electrodes, the sugar oxidation does not deactivate them.
In our recent work [5] the applicability of “conventional” voltammetric methods was investigated for diffusion coefficient measurements of a few monosaccharides, among them glucose. In those measurements, conventional-size copper electrodes as well as copper microelectrodes were used. Different chronoamperometric methods and calculations using the Randles-Sevchick equation were tested. None of these methods gave reliable values for the diffusion coefficients. The reason was obvious. The electrocatalytic oxidation of those sugar components was a complicated reaction, with multielectron exchange [6–8]. It was not diffusion-controlled.
It was expected that electrochemical time of flight (EC-TOF) measurements with a relatively long flying distance would not be affected by the slow steps in the electrode processes. Therefore, the applicability of this technique for diffusion coefficient measurements of glucose has been investigated. The present paper summarizes our results obtained in these studies. Two different microelectrodes were used as a detector: a copper microelectrode and an amperometric glucose enzyme sensor. The copper electrode was used in basic media, while the enzyme sensor was used in pH 7.4 buffered media.
The term “electrochemical time of flight method” was introduced by Feldman et al. [9]. This method involves a source, from where the “flight” of the species investigated starts, a detector (microelectrode), sensing the local concentration of the species investigated, and a gap between the source and the detector. In generator-collector mode, the diffusion coefficient of redox species can be measured. Slowinska et al. [10] used micro-band electrodes, amperostatic generation, and potentiometric detection for diffusion coefficient measurements. Bard et al. [11] and Fosset et al. [12] dealt with the theory of the EC-TOF measurements. Recently Amatore [13] investigated theoretically the EC-TOF method, considering the geometry of a flat-form cell containing two parallel micro-band electrodes.
In the time-of-flight studies of Mosbach et al. [14], K4[Fe(CN)6] and ferrocene solutions were injected using a microdispenser close to the collector microelectrode. The position of the microdispenser was set with high precision.
The diffusion coefficient of Br- ions in solid AgBr films was measured by Mirkin et al. [15] using the EC-TOF method. Potential pulses of 50–200 ms were applied on a Ag/AgBr disc generator electrode, and the time necessary for the ions to cross the gap between the generator and the detector electrode was measured. The diffusion coefficient values in AgBr and in water were compared.
In our early experiments [16] in the laboratory of Ralph Adams, we measured the migration rate of different neurotransmitter-related molecules in vivo in the brain tissues of anesthetized rats. In those experiments a microdroplet of the solution of the species was pressure-ejected and the concentration transient was detected by a carbon microelectrode positioned in the close vicinity of the ejector. Uncertainty of the gap size measurements could introduce errors.
The scanning electrochemical microscope (SECM) introduced by Bard et al. [17, 18] can accurately change the distance between the generator and the collector units; therefore, EC-TOF measurements with several flight distances can be made. Having several concentration-time transients recorded at different gap sizes, one can obtain more accurate data for diffusion. This was proved earlier in our work [19] on electrochemically reversible species. Amperostatic pulses were used for generation and amperometric microelectrodes were used for detection.
The micro-sized injector and the measuring tip can move axially in gels slurries or living tissues also without causing major structural damage or invasion; therefore, the method can be used for diffusion coefficient measurements in those media.
Different methods with the SECM have been used by other workers for diffusion coefficient measurements; as an example, we mention the report of Martin and Unwin [20].
Experimental
Equipment
Autolab PGSTAT12 (Eco-Chemie, Utrecht, The Netherlands) and CHI type 760C (CH Instruments, Austin, TX, USA) electrochemical workstations were used in voltammetric experiments.
The SECM apparatus used in this work was constructed in our laboratory. A detailed description has already been published [21, 22].
Electrodes
Homemade micro-sized SECM tip working electrodes were used. The copper working electrodes were prepared from high-purity copper wires (diameter 20 μm) obtained from GoodFellows (UK). The wires were sealed in cylindrical glass in the following way. One end of a soft glass micropipette (inner diameter 1.16 mm; Sutter, Novato, CA, USA) was sealed by a micro flame. An about 2 cm long section of copper wire (diameter 20 μm) was introduced into the lumen all the way to the sealed end. With use of a vacuum and an electric heating coil, part of the microwire was sealed in the glass column (about 1 cm long). Copper wire was inserted into the lumen of the micropipette and soldered to the microwire to serve as an electric contact. Then the tip was formed by sandpaper filing, beveling, and finally polishing with Al2O3 powder. A pointy tip with a glass to metal diameter ratio of about 6:1 at the tip was produced. The surfaces of the disc electrodes were polished with wet alumina polishing powder. Polishing powder of 1-, 0.3-, and 0.05-μm diameter (Micropolish II, Buehler, USA) was used sequentially. Sometimes the copper electrodes were renewed by wet-polishing their surface with 0.05-μm-diameter alumina polishing powder.
The pointy platinum microdisc electrode used as a base sensor of the glucose enzyme electrode was prepared similarly. In this case platinum wire (20-μm diameter; GoodFellows UK) was sealed in the glass micropipette.
Platinum wire of 1-mm diameter served as the auxiliary electrode, while a silver wire was used as quasi reference one.
Enzyme immobilization
The amperometric glucose electrode was prepared by immobilizing glucose oxidase enzyme (EC 1.1.3.4, 18.5 U/mg, Sigma) on the small measuring disc of the platinum electrode. The enzyme-containing layer was made by cross-linking the enzyme using glutaric aldehyde. In the immobilization procedure, freshly prepared solutions, 1 μl glucose oxidase solution of 3.2–4.7-U activity and 1 μl 4.5% glutaric aldehyde solution, both in pH 7.4 phosphate buffer, were mixed and the mixture was quickly spread on the tip of the working electrode with a microsyringe. The film was allowed to reticulate for 1 h at room temperature.
Finally, a thin polyurethane, Tecoflex SG-80A (Thermedics, Woburn, MA, USA), film as a protective external layer was prepared by spreading 1 μl very dilute polyurethane solution over the whole sensor surface. To prepare this spreading solution, 1 g urethane was dissolved in 100 ml tetrahydrofuran. This solution was diluted 500 times with tetrahydrofuran. The total area of the electrode tip (platinum disc diameter 20 μm, total diameter with the glass shield was about 140 μm) was about 1.5 × 10−2 mm2. The polyurethane film thickness was estimated to be 1.2 μm.
All measurements with the copper electrode were made in 100 mM sodium hydroxide background solution. Our earlier investigations [23] proved the advantage of using it in voltammetric sugar analysis.
Micropipettes with a typical tip diameter of 0.2–1 μm were used for pressure ejection of nanodroplets of the glucose solution. The pipettes were pulled with Sutter horizontal puller (Sutter Instrument. Novato, CA, USA). The pipettes were connected to a Rheodyne type 9125i loop injector (Rheodyne, CA, USA). A cylinder containing high-purity nitrogen was connected to the loop injector. The droplet was delivered by manually changing the inject/load position of the injector, which caused a short pressure pulse in the pipette.
For voltammetric measurements to test the electrodes, small-volume beakers served as measurement cells, while cylindrical Plexiglas cells were prepared for the SECM measurements. The bottom of the cell was made of a rubber plug. The copper microelectrode was introduced through a hole in the bottom, while the micropipette was held by the holder fixed to the vertical positioning linear motor of the microscope.
Analytical-grade D-(+)-glucose was purchased from Sigma-Aldrich. Freshly prepared 0.1 M sodium hydroxide solution or pH 7.4 phosphate (Sörensen) buffer solution was used as the background electrolyte. All the solutions were prepared with deionized water (specific conductivity less than 0.5 μS cm−1). SECM time of flight (SECM-TOF) diffusion coefficient measurements were made in 0.5% agarose gel prepared with buffer solution and in different sand slurries made with buffer or sodium hydroxide solution.
Measuring with the SECM-TOF method
The schematic outline of the measurement setup is shown in Fig. 1 It can be seen that the detector, the micro-sized working electrode (electrocatalytic copper microdisc or amperometric glucose enzyme electrode), and the sample source (micropipette filled with the glucose solution) were set close to each other. A pressure-delivery Tygon tube connects the output side of the loop injector to the droplet-delivery micropipette. Nitrogen gas from a cylinder produced the overpressure pulse. The loop injector was operated manually.

The scanning electrochemical microscope time of flight experimental arrangement
The “flight distance” between the source and the detector is represented by d. Accurate setting of the initial distance (d i) cannot be done on the micrometer scale. However, the positioning device of the microscope can change the distance with high precision, making a known number (n) of steps of known length (Δx). In this way, several flight distances (d = d i±nΔx) can be set and used for measurements. To carry out the diffusion coefficient determination, a very small droplet of the solution of the compound studied is delivered from the pipette tip. If convection is avoided, the species introduced spherically diffuse into the bulk of the medium. If the shielding effect of the stem of the pointy micropipette can be neglected, the concentration of the species at distance d from the infinitesimal-size droplet at time t (C d,t ) is given as [24]
where M is the amount of the species in the droplet and D is the diffusion coefficient. At a location in the medium a certain distance (d) from the source, the concentration reaches a maximum as the diffusing species are passing by. The time period between the droplet delivery and the time of the concentration maximum (Δt max) can be expressed easily by finding the maximum of the C d -t dependence. Performing the calculation, we get a very simple expression (see details in the Appendix):
This indicates that without knowing the amount of the material delivered, we can obtain the diffusion coefficient from the d and Δt max data pair:
In our earlier [16] experimental setup, we used an optical microscope for measurement of the preset d value and very small droplets were delivered. It became clear, however, that the reliability of the EC-TOF method could be greatly improved by using the SECM. The applicability of the SECM-TOF method was tested earlier [19]. In that work, mostly electrochemically reversible or semireversible species were studied and electrode reactions were used for delivery.
The SECM-TOF method using Eq. 3 can give reliable results only if the following conditions are met:
-
1.
The radius of the droplet injected is negligible compared with the flight distance.
-
2.
No convection is involved in the transport (density differences, fast droplet ejection, and vibrations must be avoided).
-
3.
No substantial obstruction in the spherical diffusion field shows up.
-
4.
The response of the detector is fast compared with the rate of concentration change.
Results and discussion
EC-TOF measurements with the copper microelectrode
In time-of-flight measurements of the glucose diffusion coefficient, the copper microelectrode is used as a detector in quiescent basic media in amperometric mode. The electrode potential for the amperometric detection was selected on the basis of DC voltammograms. Figure 2 shows electrode potential - current dependences obtained in 100 mM sodium hydroxide background solution (curve A) and in 1 mM glucose solution dissolved in the same background solution (curve B) (polarizing rate 100 mV/s). It can be seen that 0.65 V versus the silver wire reference electrode potential is a good selection for the amperometric glucose detection.

DC voltammograms made with 0.1 V/s scanning rate using a copper microelectrode. Curve A 100 mM sodium hydroxide background electrolyte; curve B 1 mM glucose solution in the same electrolyte
A question can arise if the copper microelectrode shows steady current in quiescent sodium hydroxide solution. To check this, 10 ml sodium hydroxide was pipetted into the measurement cell and with use of a potentiostat the copper microelectrode was polarized to 0.65 V versus the reference electrode. The amperometric current was recorded in time. Small volumes of 100 mM glucose solutions were introduced. Stirring for a short period was done after each addition. The current-time recording and the calibration curve made using the current values taken from it are shown in Fig. 3. As can be seen, the current achieves a steady value in a short time, and the steady current values indicate well the actual glucose concentration.

Glucose calibration curve obtained in quiescent 100 mM sodium hydroxide background electrolyte in amperometric mode using 0.65-V electrode potential. The inset shows the recording
In diffusion coefficient measurements the micropipette was filled up with glucose solution (100 mM, prepared with 100 mM sodium hydroxide as the solvent). Different flight distances (d = d i + nΔx) were set in the measurement cell of the SECM containing the media investigated, and the current was continuously recorded. Manually turning the handle of the loop injector to “inject” for 1–3 s resulted in a microdroplet being delivered. After each microdroplet of the glucose solution had been delivered, current-time transients appeared. Figure 4 shows current-time recordings obtained at different d values. The t = 0 time in each curve represents the starting edge of the pressure pulse. Figure 4a shows the recorded curve, while Fig. 4b shows the same transients with the i t values transformed to (i t -i r )/i t,max (where i t is the current at time t, i t,max is the current maximum, and i r is the residual current measured before the injection).

a Current-time recordings obtained at different d values using a glucose-measuring enzyme electrode tip. b The same current-time curves as shown in a after background correction and normalization. The current values are given as i = (i t -i r )/i t,max, where i r is the residual current, i t is the current at time t, and i t,max is the current maximum
To investigate the applicability of Eq. 3 for calculating the D value, we fitted the current-time curves obtained to C d -time curves calculated on the basis of Eq. 1. The fitting was done in the following way:
In a series of experiments d i and D values were determined. An i t -t recording was selected from the prerecorded ones. It is plotted as (i t -i r)/i t,max in Fig. 5 and it is called “measured transient.” Knowing the D and d = (d i + nΔx) data pairs, we generated a C t,d -t curve with the Origin 7 program using Eq. 1. This was fitted using Eq. 4 to the measured curve. To obtain the fitting constant K, the maximum of i t -i r (Δi t,d,max) was divided by the maximum of C t,d (C t,d ,max) (see Eq. 5). The calculated C t,d values were multiplied by K and plotted as a function of time. The curve obtained normalized to Δi t,d,max is the theoretical transient in Fig. 5.
where
Theoretical and measured transients were compared. Figure 5 shows the theoretical (continuous line) and measured (dotted line) i-t curves. The acceptable fitting proves the applicability of the Δt max-based D-determining EC-TOF method.

Comparison of the theoretical (continuous line) and measured (dotted line) time-of-flight transients
The diffusion coefficient we measured with the EC-TOF method is 6.3 × 10−6 cm2/s, with a standard deviation of 3.5 × 10−7 cm2/s, which is in good agreement with the value found in the literature for aqueous solutions: 6.76 × 10−6 cm2/s [25].
Measurements with the glucose enzyme sensor
It was proved that the SECM-TOF method with a copper electrode could be used well to measure the glucose diffusion coefficient. However, this could be done only if the pH of the medium was high. In neutral or acidic pH, the glucose is not electroactive in the potential range available with conventional electrodes. However, the diffusion coefficient needed to be determined in neutral media, in biological solutions or tissues; therefore, the applicability of the glucose enzyme sensor was investigated.
Before EC-TOF measurements with the glucose enzyme electrode were done, the response rate and the response stability of the microsensor had to be checked.
The slow response can influence Δt max. Similarly, if the electrode does not show a steady concentration response current in quiescent media, that would seriously limit the reliability of the D values obtained with the method.
To obtain information on the response time, the current was recorded in time at 0.65 V versus the silver wire quasi reference electrode in 10 ml intensively stirred phosphate buffer (pH 7.4). After a steady current reading had been obtained, 0.1 ml glucose solution (100 mM) was added. As can be seen in Fig. 6, the current increased and quickly achieved a steady value. The time needed to achieve 95% of the total change was used for characterization of the response time. It was in the range 5–10 s in the case of this fast concentration jump. Hopefully, in the case of slow concentration transients this will cause a small, tolerable delay.

Response curve of the glucose electrode (10 ml buffer solution, 10 µl of 0.1 M glucose solution added, electrode potential 0.65 V)
To check the response stability in quiescent solution, an experiment similar to that carried out to obtain Fig. 3 was performed. In this case 10 ml phosphate buffer background solution and a 0.65-V electrode potential were used. Ten-microliter doses of 100 mM glucose solution were added to the cell and the mixture was quickly homogenized. The current-time recording and the calibration curve obtained are shown in Fig. 7. It can be concluded that the glucose enzyme sensor in amperometric mode (with a little compromise) can be used for diffusion coefficient measurements in close to neutral media.

Current-time recording obtained in the case of addition of 100-µl doses of 100 mM glucose solution into 10 ml buffer solution. Glucose enzyme electrode, detection electrode potential 0.65 V. Inset: calibration curve
Figure 8 shows recordings obtained at different flight distances (d). t = 20 shows the starting edge of the pressure pulse. The current values in Fig. 8b are transformed to (i t -i r)/i t,max.

a Current-time recordings obtained in the case of different d values using the glucose enzyme electrode for detection in amperometric mode. Electrode potential 0.65V versus the silver quasi reference. b (i t -i r)/i t,max time recordings obtained in the case of different d values using the glucose enzyme electrode for detection in amperometric mode. Electrode potential 0.65 V versus the silver quasi reference
From two nΔx-Δt max data pairs, one can calculate the d i value:
To evaluate the data series listed in Table 1, all combinations of the nΔx-Δt max values were used for calculating d i values. The average obtained was used to calculate D values for every flight distance (d = d i + nΔx)-t max data pair using Eq. 3.
Table 1 summarizes the results obtained with enzyme electrode detection in aqueous phosphate buffer.
The pointy micropipette used for glucose droplet delivery can move vertically in gels, in tissues, in sediments, etc.; therefore, the diffusion coefficient measurements can be carried out easily. In our work 0.5% agarose gel was prepared in the measurement cell of the SECM. In preparing the diffusion medium, we dissolved 0.25 g low gelling temperature agarose (type I, low from Sigma) in 50 ml phosphate buffer by boiling. The solution was allowed to cool. When the gelling temperature had almost been achieved, the solution was poured into the cell, in which the enzyme electrode had already been inserted through the hole in the bottom plug of the cell.
The diffusion coefficient measurements were performed in a way similar to that described above. The D values obtained by setting different distances are summarized in Table 2.
Interestingly, slightly higher D values were obtained for the gel experiments. The difference, however, is not significant. In our earlier experiments [16, 19] with low-concentration agarose gels we also did not find significant differences between D values measured in gels and in solutions. Significant differences were found, however, when high molecular weight polyelectrolyte containing gels were investigated.
Discussion
The simple electrochemical methods that are generally used for diffusion coefficient determinations are not easily applicable to glucose in different media since the glucose does not show appropriate electric activity. The EC-TOF method using the SECM was developed and applied. The method is simple, and requires a relatively short measurement time. Its applicability was checked using an electrocatalytic copper oxide coated copper microelectrode as the detector as well as a micro-sized glucose enzyme sensor. The measurements with the copper electrode could be used only in basic media, while in media close to neutral pH the enzyme sensor performed well.
The method can be used in gels, tissues, and sediments. Because of the importance of glucose diffusion in different media, it is expected to be applied in different areas. The precision of the values obtained depends on the distance. The model used to obtain Eq. 3 considers spherical diffusion starting from a point source. In reality, the source is a small droplet, and there can be some convection as the droplet is formed. Furthermore, there can be some shielding of the diffusion route by the stem of the pipette or the electrode body. The longer the distance, the more closely the experimental conditions fit to the theoretical ones, i.e., to the model that resulted in Eq. 1. The behavior is less affected by the size of the droplet, by the convection caused by the droplet forming, and by the shielding. However, when the distance is too long, the concentration at the detector can become very small, so the uncertainty of the location of i max or t max on the recordings can lower the precision. Accordingly, a compromise has to be made when selecting the range of flight distance values.
The diffusion coefficient values obtained in our measurements, compared with the literature values, can be accepted especially knowing that the accuracy of other methods is usually not excellent. The EC-TOF method proved to be a good application field for the quite powerful SECM technique.
Conclusion
The voltammetric response of glucose in the potential window available in the case of the generally used working electrodes does not allow direct application of different simple electrochemical diffusion coefficient measurement methods. The rate of glucose transport in different media, however, is of importance in different fields.
In the study presented, the application of a simple diffusion coefficient determination method was investigated. The method is based on the combined application of SECM apparatus and the EC-TOF measurement principle.
Using the SECM for the EC-TOF measurements, one can change the distance d accurately. In this way several d = (d i ± nΔx)-Δt max data pairs can be obtained. From them, d i can be calculated. It is to be hoped that performing a multidistance EC-TOF measurement series will result in more reliable D values being obtained.
In strongly basic media, an electrocatalytic copper microelectrode as a measuring tip was used, while for measurements in close to neutral solutions and agar gel, a micro-sized amperometric enzyme sensor was prepared. Owing to the special property of the SECM, acceptable diffusion coefficient values were obtained following the simple procedure, which requires a relatively short measurement time.
References
Ching CTS, Connolly P (2008) Sens Actuators B 129:30
Conde C, Silva P, Agasse A, Tavares RM, Delrot S, Gerós H (2007) Biochim Biophys Acta 1768:2801
Qutub A, Hunt CA (2005) Brain Res Rev 49:595
Zhang W, Furusaki S (2001) Biochem Eng J 9:73
Nagy L, Gyetvai G, Nagy G (2009) Electroanalysis (in press)
Torto N, Ruzgas T, Gorton L (1999) J Electroanal Chem 464:252
Ye R, Balvin P (1994) J Chromatogr A 687:141
Nagy L, Nagy G (2006) Microchem J 84:70
Feldman BJ, Feldberg SW, Murray RW (1987) J Phys Chem 91:6558
Slowinska K, Feldberg SW, Majda M (2003) J Electroanal Chem 554–555:61
Bard AJ, Crayston JA, Kittlesen GP, Shea TV, Wrighton MS (1986) Anal Chem 58:2321
Fosset B, Amatore C, Bartlet J, Wightman RM (1991) Anal Chem 63:1403
Amatore C, Sella C, Thouin L (2006) J Electroanal Chem 593:194
Mosbach M, Laurell T, Nilsson J, Csöregi E, Schuhmann W (2001) Anal Chem 73:2468
Mirkin MV, Arca M, Bard AJ (1993) J Phys Chem 97:10790
Rice ME, Gerhardt GA, Hierl PM, Nagy G, Adams RN (1985) Neuroscience 15:891
Bard AJ, Fan FRF, Kwak J, Lev O (1989) Anal Chem 61:132
Bard AJ, Mirkin MV (eds) (2001) Scanning electrochemical microscopy. Wiley, New York
Csóka B, Nagy G (2004) J Biochem Biophys Methods 61:57
Martin RD, Unwin PR (2006) J Electroanal Chem 593:19
Kovács B, Csóka B, Nagy G, Kapui I, Gyurcsányi RE, Tóth K (1999) Electroanalysis 11:349
Csóka B, Kovács B, Nagy G (2003) Electroanalysis 15:1335
Nagy L, Nagy G, Hajós P (2001) Sens Actuators B 76:493
Crank J (1975) The mathematics of diffusion, 2nd edn. Clarendon, Oxford, p 29
Lide DR (ed) (1995) CRC handbook of chemistry and physics. CRC, Boca Raton
Acknowledgements
This work was supported by Hungarian Scientific Research Fund grant T 049502 and OMFB-00479/1008 TeT research grant.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
The derivation of Eq. 2 from Eq. 1 is detailed below.
The second term has to be zero; therefore
t is actually ∆tmax, so Eq. 3 is proved:
Rights and permissions
About this article
Cite this article
Varga, Á., Gyetvai, G., Nagy, L. et al. Electrochemical time of flight method for determination of diffusion coefficients of glucose in solutions and gels. Anal Bioanal Chem 394, 1955–1963 (2009). https://doi.org/10.1007/s00216-009-2859-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-009-2859-9