
Abstract
Novel therapies are required for the treatment of metastatic renal cell carcinoma (RCC), which is associated with inoperable disease and patient death. Histone deacetylases (HDACs) are epigenetic modifiers and potential drug targets. Additional information on molecular pathways that are altered by histone deacetylase inhibitors (HDACi) in RCC cells is warranted. It should equally be delineated further which individual members of the 18 mammalian HDACs determine the survival and tumor-associated gene expression programs of such cells. Most importantly, an ongoing dispute whether HDACi promote or suppress metastasis-associated epithelial-to-mesenchymal transition (EMT) has to be resolved before HDACi are considered further as clinically relevant drugs. Here we show how HDACi affect murine and primary human RCC cells. We find that these agents induce morphological alterations resembling the metastasis-associated EMT. However, individual and proteomics-based analyses of epithelial and mesenchymal marker proteins and of EMT-associated transcription factors (EMT-TFs) reveal that HDACi do not trigger EMT. Pathway deconvolution analysis identifies reduced proliferation and apoptosis induction as key effects of HDACi. Furthermore, these drugs lead to a reduction of the cell adhesion molecule E-cadherin and of the platelet-derived growth factor receptor-β (PDGFRβ), which is a key driver of RCC metastasis formation. Accordingly, HDACi reduce the pulmonary spread of syngeneic transplanted renal carcinoma cells in mice. Specific genetic elimination of the histone deacetylases HDAC1/HDAC2 reflects the effects of pharmacological HDAC inhibition regarding growth suppression, apoptosis, and the downregulation of E-cadherin and PDGFRβ. Thus, these epigenetic modifiers are non-redundant gatekeepers of cell fate and precise pharmacological targets.







Similar content being viewed by others

Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.References
Beyer M, Kiweler N, Mahboobi S, Krämer OH (2017) How to distinguish between the activity of HDAC1-3 and HDAC6 with Western Blot. Methods Mol Biol 1510:355–364. https://doi.org/10.1007/978-1-4939-6527-4_26
Blaheta RA, Michaelis M, Driever PH, Cinatl J Jr (2005) Evolving anticancer drug valproic acid: insights into the mechanism and clinical studies. Med Res Rev 25:383–397. https://doi.org/10.1002/med.20027
Brabletz T (2012) To differentiate or not-routes towards metastasis. Nat Rev Cancer 12:425–436. https://doi.org/10.1038/nrc3265
Bradner JE et al (2010) Chemical genetic strategy identifies histone deacetylase 1 (HDAC1) and HDAC2 as therapeutic targets in sickle cell disease. Proc Natl Acad Sci USA 107:12617–12622. https://doi.org/10.1073/pnas.1006774107
Carew RM, Wang B, Kantharidis P (2012) The role of EMT in renal fibrosis. Cell Tissue Res 347:103–116. https://doi.org/10.1007/s00441-011-1227-1
Cha TL et al (2009) Dual degradation of aurora A and B kinases by the histone deacetylase inhibitor LBH589 induces G2-M arrest and apoptosis of renal cancer cells. Clin Cancer Res 15:840–850. https://doi.org/10.1158/1078-0432.ccr-08-1918
Christmann M, Kaina B (2013) Transcriptional regulation of human DNA repair genes following genotoxic stress: trigger mechanisms, inducible responses and genotoxic adaptation. Nucleic Acids Res 41:8403–8420. https://doi.org/10.1093/nar/gkt635
Dejung M et al (2016) Quantitative proteomics uncovers novel factors involved in developmental differentiation of trypanosoma brucei. PLoS Pathog 12:e1005439. https://doi.org/10.1371/journal.ppat.1005439
Falcon S, Gentleman R (2007) Using GOstats to test gene lists for GO term association. Bioinformatics 23:257–258. https://doi.org/10.1093/bioinformatics/btl567
Falkenberg KJ, Johnstone RW (2014) Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov 13:673–691. https://doi.org/10.1038/nrd4360
Fischer KR et al (2015) Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527:472–476. https://doi.org/10.1038/nature15748
Fritzsche FR et al (2008) Class I histone deacetylases 1, 2 and 3 are highly expressed in renal cell cancer. BMC Cancer 8:381–381. https://doi.org/10.1186/1471-2407-8-381
Giudice FS, Pinto DS Jr, Nor JE, Squarize CH, Castilho RM (2013) Inhibition of histone deacetylase impacts cancer stem cells and induces epithelial–mesenchyme transition of head and neck cancer. PLoS One 8:e58672. https://doi.org/10.1371/journal.pone.0058672
Göttlicher M et al (2001) Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 20:6969–6978. https://doi.org/10.1093/emboj/20.24.6969
Han HJ et al (2014) Suppression of E-cadherin mediates gallotannin induced apoptosis in Hep G2 hepatocellular carcinoma cells. Int J Biol Sci 10:490–499. https://doi.org/10.7150/ijbs.7495
Jiang GM et al (2013) Histone deacetylase inhibitor induction of epithelial–mesenchymal transitions via up-regulation of Snail facilitates cancer progression. Biochim Biophys Acta 1833:663–671. https://doi.org/10.1016/j.bbamcr.2012.12.002
Jones J, Juengel E, Mickuckyte A, Hudak L, Wedel S, Jonas D, Blaheta RA (2009a) The histone deacetylase inhibitor valproic acid alters growth properties of renal cell carcinoma in vitro and in vivo. J Cell Mol Med 13:2376–2385. https://doi.org/10.1111/j.1582-4934.2008.00436.x
Jones J et al (2009b) Valproic acid blocks adhesion of renal cell carcinoma cells to endothelium and extracellular matrix. J Cell Mol Med 13:2342–2352. https://doi.org/10.1111/j.1582-4934.2008.00603.x
Juengel E et al (2011) Alterations of the gene expression profile in renal cell carcinoma after treatment with the histone deacetylase-inhibitor valproic acid and interferon-alpha. World J Urol 29:779–786. https://doi.org/10.1007/s00345-010-0582-y
Juengel E et al (2014) HDAC-inhibition counteracts everolimus resistance in renal cell carcinoma in vitro by diminishing cdk2 and cyclin A. Mol Cancer 13:152. https://doi.org/10.1186/1476-4598-13-152
Kanao K, Mikami S, Mizuno R, Shinojima T, Murai M, Oya M (2008) Decreased acetylation of histone H3 in renal cell carcinoma: a potential target of histone deacetylase inhibitors. J Urol 180:1131–1136. https://doi.org/10.1016/j.juro.2008.04.136
Keller SH, Nigam SK (2003) Biochemical processing of E-cadherin under cellular stress. Biochem Biophys Res Commun 307:215–223
Korfei M et al (2015) Aberrant expression and activity of histone deacetylases in sporadic idiopathic pulmonary fibrosis. Thorax 70:1022–1032. https://doi.org/10.1136/thoraxjnl-2014-206411
Lagger G et al (2002) Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J 21:2672–2681. https://doi.org/10.1093/emboj/21.11.2672
Li Y, Seto E (2016) HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harbor Perspectives in Medicine 6 https://doi.org/10.1101/cshperspect.a026831
Li Z, Zhu WG (2014) Targeting histone deacetylases for cancer therapy: from molecular mechanisms to clinical implications. Int J Biol Sci 10:757–770. https://doi.org/10.7150/ijbs.9067
Mahalingam D et al (2010) Vorinostat enhances the activity of temsirolimus in renal cell carcinoma through suppression of survivin levels. Clin Cancer Res 16:141–153. https://doi.org/10.1158/1078-0432.CCR-09-1385
Maurer-Gebhard M, Schmidt M, Azemar M, Stocklin E, Wels W, Groner B (1999) A novel animal model for the evaluation of the efficacy of drugs directed against the ErbB2 receptor on metastasis formation. Hybridoma 18:69–75. https://doi.org/10.1089/hyb.1999.18.69
Meidhof S et al (2015) ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol Med 7:831–847. https://doi.org/10.15252/emmm.201404396
Menegola E, Di Renzo F, Broccia ML, Prudenziati M, Minucci S, Massa V, Giavini E (2005) Inhibition of histone deacetylase activity on specific embryonic tissues as a new mechanism for teratogenicity. Birth Defects Res B Dev Reprod Toxicol 74:392–398. https://doi.org/10.1002/bdrb.20053
Mikami S et al (2016) Recent advances in renal cell carcinoma from a pathological point of view. Pathol Int 66:481–490. https://doi.org/10.1111/pin.12433
Nieto MA, Huang RY, Jackson RA, Thiery JP (2016) Emt: 2016. Cell 166:21–45. https://doi.org/10.1016/j.cell.2016.06.028
Nikolova T, Kiweler N, Krämer OH (2017) Interstrand crosslink repair as a target for HDAC inhibition. Trends Pharmacol Sci. https://doi.org/10.1016/j.tips.2017.05.009
Park KC et al (2015) The novel histone deacetylase inhibitor, N-hydroxy-7-(2-naphthylthio) hepatonomide, exhibits potent antitumor activity due to cytochrome-c-release-mediated apoptosis in renal cell carcinoma cells. BMC Cancer 15:19. https://doi.org/10.1186/s12885-014-1003-1
Pili R et al (2017) Combination of the histone deacetylase inhibitor vorinostat with bevacizumab in patients with clear-cell renal cell carcinoma: a multicentre, single-arm phase I/II clinical trial. Br J Cancer 116:874–883. https://doi.org/10.1038/bjc.2017.33
Piva F et al (2016) Epithelial to mesenchymal transition in renal cell carcinoma: implications for cancer therapy. Mol Diagn Ther 20:111–117. https://doi.org/10.1007/s40291-016-0192-5
Poreba M, Strozyk A, Salvesen GS, Drag M (2013) Caspase substrates and inhibitors. Cold Spring Harb Perspect Biol 5:a008680. https://doi.org/10.1101/cshperspect.a008680
Ramakrishnan S, Pili R (2013) Histone deacetylase inhibitors and epigenetic modifications as a novel strategy in renal cell carcinoma. Cancer J 19:333–340. https://doi.org/10.1097/PPO.0b013e3182a09e07
Rhodes LV et al (2014) Suppression of triple-negative breast cancer metastasis by pan-DAC inhibitor panobinostat via inhibition of ZEB family of EMT master regulators. Breast Cancer Res Treat 145:593–604. https://doi.org/10.1007/s10549-014-2979-6
Roy SS et al (2014) Significance of PELP1/HDAC2/miR-200 regulatory network in EMT and metastasis of breast cancer. Oncogene 33:3707–3716. https://doi.org/10.1038/onc.2013.332
Ruscetti M et al (2016) HDAC inhibition impedes epithelial–mesenchymal plasticity and suppresses metastatic, castration-resistant prostate cancer. Oncogene 35:3781–3795. https://doi.org/10.1038/onc.2015.444
Schech A, Kazi A, Yu S, Shah P, Sabnis G (2015) Histone deacetylase Inhibitor entinostat inhibits tumor-initiating cells in triple-negative breast cancer cells. Mol Cancer Ther 14:1848–1857. https://doi.org/10.1158/1535-7163.mct-14-0778
Shibue T, Weinberg RA (2017) EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol Adv Online Publ. https://doi.org/10.1038/nrclinonc.2017.44
Society AC (2017) Cancer Facts & Figs. 2017. https://old.cancer.org/acs/groups/content/@editorial/documents/document/acspc-048738.pdf. Accessed 13 June 2017
Srivastava RK, Kurzrock R, Shankar S (2010) MS-275 sensitizes TRAIL-resistant breast cancer cells, inhibits angiogenesis and metastasis, and reverses epithelial–mesenchymal transition in vivo. Mol Cancer Ther 9:3254–3266. https://doi.org/10.1158/1535-7163.mct-10-0582
Tang HM et al (2016) An epithelial marker promoter induction screen identifies histone deacetylase inhibitors to restore epithelial differentiation and abolishes anchorage independence growth in cancers. Cell Death Discov 2:16041. https://doi.org/10.1038/cddiscovery.2016.41
Voon DC, Huang RY, Jackson RA, Thiery JP (2017) The EMT spectrum and therapeutic opportunities. Mol Oncol 11:878–891. https://doi.org/10.1002/1878-0261.12082
Wu S, Luo Z, Yu PJ, Xie H, He YW (2016a) Suberoylanilide hydroxamic acid (SAHA) promotes the epithelial mesenchymal transition of triple negative breast cancer cells via HDAC8/FOXA1 signals. Biol Chem 397:75–83. https://doi.org/10.1515/hsz-2015-0215
Wu Y, Lyu H, Liu H, Shi X, Song Y, Liu B (2016b) Downregulation of the long noncoding RNA GAS5-AS1 contributes to tumor metastasis in non-small cell lung cancer. Sci Rep 6:31093. https://doi.org/10.1038/srep31093
Xu L, Tong R, Cochran DM, Jain RK (2005) Blocking platelet-derived growth factor-D/platelet-derived growth factor receptor beta signaling inhibits human renal cell carcinoma progression in an orthotopic mouse model. Cancer Res 65:5711–5719. https://doi.org/10.1158/0008-5472.CAN-04-4313
Yamada T, Horinaka M, Shinnoh M, Yoshioka T, Miki T, Sakai T (2013) A novel HDAC inhibitor OBP-801 and a PI3K inhibitor LY294002 synergistically induce apoptosis via the suppression of survivin and XIAP in renal cell carcinoma. Int J Oncol 43:1080–1086. https://doi.org/10.3892/ijo.2013.2042
Yao X, Ireland SK, Pham T, Temple B, Chen R, Raj MH, Biliran H (2014) TLE1 promotes EMT in A549 lung cancer cells through suppression of E-cadherin. Biochem Biophys Res Commun 455:277–284. https://doi.org/10.1016/j.bbrc.2014.11.007
Ye Y, Xiao Y, Wang W, Yearsley K, Gao JX, Shetuni B, Barsky SH (2010) ERalpha signaling through slug regulates E-cadherin and EMT. Oncogene 29:1451–1462. https://doi.org/10.1038/onc.2009.433
Yoo CB, Yun SM, Jo C, Koh YH (2012) gamma-Secretase-dependent cleavage of E-cadherin by staurosporine in breast cancer cells. Cell Commun Adhes 19:11–16. https://doi.org/10.3109/15419061.2012.665969
Zeisberg M, Neilson EG (2009) Biomarkers for epithelial–mesenchymal transitions. J Clin Investig 119:1429–1437. https://doi.org/10.1172/JCI36183
Zheng X et al (2015) Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527:525–530. https://doi.org/10.1038/nature16064
Zhou X et al (2014) Harnessing the p53-PUMA axis to overcome DNA damage resistance in renal cell carcinoma. Neoplasia 16:1028–1035. https://doi.org/10.1016/j.neo.2014.09.012
Zibelman M et al (2015) Phase I study of the mTOR inhibitor ridaforolimus and the HDAC inhibitor vorinostat in advanced renal cell carcinoma and other solid tumors. Investig New Drugs 33:1040–1047
Zimmermann S et al (2007) Reduced body size and decreased intestinal tumor rates in HDAC2-mutant mice. Can Res 67:9047–9054. https://doi.org/10.1158/0008-5472.can-07-0312
Acknowledgements
The Wilhelm Sander-Stiftung (2010.078 to O.H.K) supported the major part of this work. The laboratory of O.H.K is additionally supported by the Deutsche Forschungsgemeinschaft (KR2291/4-1, KR2291/5-1 and KR2291/7-1 to O.H.K), the Deutsche Krebshilfe (110909 to O.H.K; German Cancer Aid) and intramural funding (University Medical Center Mainz and Naturwissenschaftlich-medizinisches Forschungszentrum Mainz, NMFZ). We thank Franziska Müller, Dagmar Faust and Tina Brachetti for excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
204_2018_2229_MOESM1_ESM.tif
Supplementary material 1 Morphological changes in Renca cells upon HDACi treatment. Renca cells were treated with the indicated concentrations of VPA (mM) and MS-275 (µM) for 24–48 h and analyzed for morphological changes with phase-change light microscopy. Images are representative for four independent experiments. Shown are the same, complete pictures of Fig. 1B. (TIF 25509 KB)
204_2018_2229_MOESM2_ESM.tif
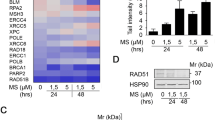
Supplementary material 2 Regulation of EMT associated factors. (A) Renca cells were treated with the indicated concentrations of MS-275 (µM) for 24–48 h. Four independent replicates were analyzed for global protein expression by label-free quantitation (LFQ) via mass spectrometry. Heatmap lists changes in LFQ expression levels of the indicated proteins. (B) Renca cells were treated with the indicated concentrations of MS-275 (µM) for 24–48 h. Four independent replicates were analyzed for global protein expression by label-free quantitation (LFQ) via mass spectrometry. Heatmap lists changes in LFQ expression levels of the indicated proteins. (C) Renca cells were treated with 1.5 and 5 µM MS-275 for 24–48 h and independent triplicates were analyzed for quantitative mRNA expression of Cdh1 encoding E-cadherin by qPCR. Graph shows mean ± SD (n = 3); one-way ANOVA; Dunnett multiple comparisons test; **P<0.01) (TIF 25509 KB)
204_2018_2229_MOESM3_ESM.tif
Supplementary material 3 HDACi reduce proliferation of Renca cells in vitro. (A) Graph depicts percentage of cells in S phase and G2 phase of Fig. 4c. (n = 4; one-way ANOVA; Dunnett multiple comparisons test; *P <0.05, ***P <0.001, ****P <0.0001). (B) Representative pictures for quantification of Giemsa staining in Fig. 4 F (TIF 25509 KB)
204_2018_2229_MOESM4_ESM.tif
Supplementary material 4 Quantification of HDAC1 and HDAC2 expression and morphological changes in Renca cells transfected with siRNAs against HDAC1 and HDAC2. (A) Densitometric analysis of HDAC1 levels detected by Western blot in Fig. 7a. Data were normalized to the respective loading controls. Results display relative amount of HDAC1 as mean ± SD (n = 3). (B) Densitometric analysis of HDAC2 levels detected by Western blot in Fig. 7a. Data were normalized to the respective loading controls. Results display relative amount of HDAC2 as mean ± SD (n = 3). (C) Cells were transfected with siRNAs against HDAC1 and HDAC2 and analyzed for morphological changes with phase-change light microscopy. Images are representative for three independent experiments. Shown are the same, complete pictures of Fig. 7b (TIF 25509 KB)
204_2018_2229_MOESM5_ESM.pdf
Supplementary material 5 Table S1 Primer sequences for qPCR analyses shown in Fig. 3a, b and Supplemental Figure S2C (PDF 19 KB)
204_2018_2229_MOESM8_ESM.xlsx
Supplementary material 6 Table S2 Complete raw data set acquired by mass-spectrometry showing global protein expression (as log2 LFQ values) of Renca cells in response to 24–48 h treatment with 1.5 and 5 µM MS-275. (XLSX 107 KB)
Rights and permissions
About this article

Cite this article
Kiweler, N., Brill, B., Wirth, M. et al. The histone deacetylases HDAC1 and HDAC2 are required for the growth and survival of renal carcinoma cells. Arch Toxicol 92, 2227–2243 (2018). https://doi.org/10.1007/s00204-018-2229-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-018-2229-5