
Abstract
Aims/hypothesis
High sodium (HS) effects on hypertension are well established. Recent evidence implicates a relationship between HS intake and insulin resistance, even in the absence of hypertension. The aim of the current study was to determine whether loss of the vascular actions of insulin may be the driving factor linking HS intake to insulin resistance.
Methods
Sprague Dawley rats were fed a control (0.31% wt/wt NaCl) or HS (8.00% wt/wt NaCl) diet for 4 weeks and subjected to euglycaemic–hyperinsulinaemic clamp (10 mU min−1 kg−1) or constant-flow pump-perfused hindlimb studies following an overnight fast. A separate group of HS rats was given quinapril during the dietary intervention and subjected to euglycaemic–hyperinsulinaemic clamp as above.
Results
HS intake had no effect on body weight or fat mass or on fasting glucose, insulin, endothelin-1 or NEFA concentrations. However, HS impaired whole body and skeletal muscle glucose uptake, in addition to a loss of insulin-stimulated microvascular recruitment. These effects were present despite enhanced insulin signalling (Akt) in both liver and skeletal muscle. Constant-flow pump-perfused hindlimb experiments revealed normal insulin-stimulated myocyte glucose uptake in HS-fed rats. Quinapril treatment restored insulin-mediated microvascular recruitment and muscle glucose uptake in vivo.
Conclusions/interpretation
HS-induced insulin resistance is driven by impaired microvascular responsiveness to insulin, and is not due to metabolic or signalling defects within myocytes or liver. These results imply that reducing sodium intake may be important not only for management of hypertension but also for insulin resistance, and highlight the vasculature as a potential therapeutic target in the prevention of insulin resistance.
Similar content being viewed by others
Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.Avoid common mistakes on your manuscript.
Introduction
Evidence is increasing in both humans and experimental animals that besides causing hypertension high sodium (HS) intake leads to insulin resistance [1–4]. Elevated BP responses to HS intake appear to be heterogeneous in nature [1, 2, 5, 6] with certain animal models and indeed humans displaying insulin resistance in the absence of hypertension [1, 2]. Thus, the mechanisms by which elevated sodium intake causes insulin resistance appear to have origins that are separate from or may precede those that cause hypertension.
An intriguing aspect of HS-induced insulin resistance is that it develops despite a paradoxical increase in insulin signalling in muscle and liver cells [2, 3]. Administration of insulin sensitisers such as pioglitazone [2], which have been shown to improve insulin sensitivity in fat-fed [7] and obese animals [8], does not prevent HS-induced insulin resistance. Therefore, the mechanisms responsible for sodium-derived insulin resistance are likely to differ from those attributed to other models of insulin resistance. Despite enhancement of hepatic and myocyte insulin signalling, vascular endothelial insulin signalling, leading to endothelial nitric oxide synthase (NOS) activation and nitric oxide (NO) production, is downregulated during HS intake [9, 10]. Thus, one factor that may be involved in the pathogenesis of sodium-derived insulin resistance is the loss of vascular responsiveness to insulin.
We have demonstrated that insulin possesses important vascular actions in skeletal muscle [11–13]. These actions include increasing both total blood flow to skeletal muscle [14] and flow through the microvascular network (microvascular recruitment) supplying myocytes [11, 12]. This microvascular action of insulin improves the delivery of insulin and glucose to myocytes and contributes approximately 40% of insulin-stimulated glucose disposal in vivo [15, 16]. NOS inhibition blocks most, if not all, insulin-stimulated microvascular recruitment in muscle demonstrating that this action is NO-dependent [16]. In healthy humans [17] and experimental animals [18–20] insulin increases microvascular recruitment. This action is markedly attenuated during states of insulin resistance (high-fat diet/obesity) and is closely associated with decreased muscle glucose uptake in vivo [17, 21–23].
Numerous studies highlight structural and functional vascular alterations following HS intake [5, 6, 9, 10, 24, 25]. These studies provide an important link between vascular dysfunction and hypertension during HS intake. However, the role of the vasculature in the development of sodium-induced insulin resistance has not been previously investigated. Thus, the primary aim of the current study was to determine whether or not HS-induced insulin resistance develops as a consequence of impaired vascular function in skeletal muscle. Since sodium-induced vascular dysfunction is likely to involve deleterious angiotensin II (AngII) activity [6, 9], a secondary aim of the present study was to investigate whether or not the development of sodium-induced insulin resistance can be prevented using the ACE inhibitor quinapril.
Methods
Animal care
Male Sprague Dawley rats (5 weeks old) obtained from the University of Tasmania Central Animal Facility (Hobart, TAS, Australia) were provided water and nutrient-matched semi-purified diets (AIN-93G, Specialty Feeds, Glen Forest, WA, Australia) ad libitum. Rats were split into two groups and given control (0.31% wt/wt NaCl; CTRL) or HS (8.00% wt/wt NaCl) diets for 4 weeks. Rats were housed at 21 ± 2°C with a 12 h–12 h light–dark cycle. All experimental procedures were approved by the University of Tasmania Animal Ethics Committee and performed in accordance with the Australian Code for the Care and Use of Animals for Scientific Purposes 2013, 8th Edition. Overnight fasted rats were subjected to either in vivo euglycaemic–hyperinsulinaemic clamps (study 1; insulin clamp) or ex vivo isolated constant-flow hindlimb perfusions (study 2).
A second group of HS rats was treated with quinapril (HS + Q; Sigma Aldrich, St Louis, MO, USA) for the duration of the dietary intervention. Quinapril was administered in drinking water at a dose of 1 mg kg−1 day−1, which has been reported to almost completely inhibit ACE activity in the plasma and vasculature of rats [26]. Overnight fasted HS + Q rats were subjected to insulin clamp experiments (study 3).
Study 1: euglycaemic–hyperinsulinaemic clamp
Surgery and protocol
Rats were anaesthetised using an intraperitoneal injection of pentobarbital sodium (50 mg/kg body weight) and their body temperature was maintained at 37°C. Surgery was performed as previously outlined [23]. Following surgery, an infusion of insulin (10 mU kg−1 min−1; Humulin, Eli Lilly, West Ryde, NSW, Australia) or saline (10 μl/min) was initiated (Fig. 1a).

Experimental protocols. (a) Studies 1 and 3: euglycaemic–hyperinsulinaemic clamp. A 3-[3H]-d-glucose bolus was administered prior to co-infusion of 3-[3H]-d-glucose with either saline (10 μl/min) or insulin. Arterial plasma was sampled as indicated and GIR was adjusted accordingly to maintain euglycaemia. Following an allopurinol bolus, 1-MX infusion was initiated and maintained throughout the experiment. At 75 min, a 2DG bolus was administered and arterial plasma was collected to determine the 2DG clearance. At 120 min, arterial and femoral vein plasma (A-V), as well as various tissue samples were collected and stored at −80°C. (b) Study 2: isolated constant-flow hindlimb perfusion. Saline or insulin was infused for 60 min. At 30 min, an infusion of 2DG was initiated. At 60 min, calf muscle was quick-frozen in liquid nitrogen and stored at −80°C
Rates of whole body glucose appearance (Ra) and disappearance (Rd) were determined using 3-[3H]-d-glucose (3.7 kBq/min, PerkinElmer, Downers Grove, IL, USA) incorporated in saline/insulin solutions (Fig. 1a) and calculated as previously described [27]. Rate of muscle glucose uptake (R′g) was determined using 2-deoxy-d-[1-14C]-glucose (2DG; 740 kBq; PerkinElmer) as previously described [23, 28]. Ser473-Akt phosphorylation was determined using western blot as previously outlined [23].
Plasma biochemistry
Plasma glucose and lactate were determined using a glucose analyser (YSI2300, Yellow Springs Instruments, Yellow Springs, OH, USA). Baseline and post-clamp plasma insulin (Mercodia AB, Uppsala, Sweden) and endothelin-1 (ET1; Biomedica, Vienna, Austria) concentrations were determined by ELISA. Plasma NEFA concentrations were determined using a commercial kit (NEFA-C, Wako Pure Chemical Industries, Osaka, Japan).
Microvascular recruitment
Microvascular recruitment in skeletal muscle was assessed using the 1-methyl xanthine (1-MX; Sigma Aldrich) method as previously published [11, 12, 16, 18, 21–23, 29, 30]. A bolus of allopurinol (10 μmol/kg; Sigma Aldrich) was administered 5 min prior to 1-MX infusion to partially inhibit the activity of xanthine oxidase thus reducing the arterial concentration of 1-MX required for saturation. This ensures that 1-MX metabolism reflects recruitment (surface area of exposure to xanthine oxidase) and not net flow rate. 1-MX (0.4 mg min−1 kg−1) was infused at 60−120 min of experiments (Fig. 1a). At 120 min, plasma 1-MX and oxypurinol concentrations were determined by reverse-phase HPLC as described [11]. Hindlimb 1-MX metabolism was determined from arteriovenous plasma differences multiplied by plasma femoral artery blood flow (FBF). Xanthine oxidase activity in skeletal muscle was determined as previously described [31].
Study 2: isolated constant-flow hindlimb perfusion
The vasculature in this preparation is fully vasodilated, thus making it ideal for studying insulin-mediated muscle glucose uptake in the absence of microvascular recruitment. We have previously used this method to demonstrate myocyte-specific insulin resistance in obese Zucker [32] and fat-fed [23] rats.
Surgical procedure
Overnight fasted rats were anaesthetised with a near-lethal intraperitoneal injection of pentobarbital sodium (60 mg/kg body weight). Surgery was performed to isolate one hindlimb as previously described [33, 34]. Immediately following surgery the animals were killed with an intracardiac injection of pentobarbital sodium and a single hindlimb perfusion was carried out as previously described [23].
Skeletal muscle glucose uptake
Saline or insulin (1.5 nmol/l final concentration) was infused continuously for 60 min (Fig. 1b). Radiolabelled glucose (2DG; 740 kBq/ml, PerkinElmer) was infused during the last 30 min to determine muscle glucose uptake as previously described [28]. The calf-muscle group (gastrocnemius, plantaris and soleus) was excised at 60 min, frozen in liquid nitrogen and kept at −80°C.
Study 3: euglycaemic–hyperinsulinaemic clamp in HS + Q rats
Animals were fasted overnight, anaesthetised and subjected to insulin clamp experiments. Muscle and liver insulin sensitivity as well as vascular measures were determined as in study 1.
Statistical analysis
All data are presented as mean ± SEM. Statistics were performed using SigmaPlot (Version 11; Systat Software, San Jose, CA, USA). Comparisons between groups were made using one-way ANOVA. Comparisons of time-series measurements in groups were performed by two-way repeated-measures ANOVA. When a significant difference (p < 0.05) was detected, pairwise comparisons by Student–Newman–Keuls was used to assess treatment differences.
Results
Physical and biochemical characteristics
No difference in body weight or epididymal fat mass was detected between the groups (Table 1). Fasting plasma concentrations of glucose, insulin, lactate, ET1 and NEFA were similar between the groups. During anaesthesia, baseline mean arterial pressure (MAP) was not different between the groups and remained constant over the course of the experiment (data not shown). Basal FBF was 45% lower in HS rats, translating to a 95% increase in femoral vascular resistance (FVR).
Study 1: euglycaemic–hyperinsulinaemic clamp
Whole body glucose kinetics
Glucose concentrations were clamped to the respective fasting level in both groups. Thus, no significant deviations in plasma glucose occurred over the course of the experiment in either group (Fig. 2a). Following insulin clamps, the plasma insulin concentration was 15% higher in HS vs CTRL rats (1,300 ± 90 vs 1,500 ± 90 pmol/l; p = 0.029). Despite this finding, the glucose infusion rate (GIR) in HS-fed rats was significantly reduced from 30 min (Fig. 2b; p = 0.023) culminating in a 20% difference at 120 min (19.7 ± 0.5 vs 16.1 ± 0.7 mg kg−1 min−1, p < 0.001). Plasma ET1 concentrations remained similar to baseline in both CTRL (2.73 ± 0.28 vs 2.72 ± 0.31 pmol/l, p = 0.99) and HS (2.48 ± 0.63 vs 3.14 ± 0.37 pmol/l, p = 0.243) groups, and were not different from each other following insulin infusion (p = 0.469).

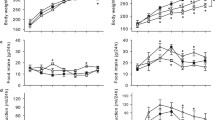
Whole body and skeletal muscle glucose kinetics in vivo. Time courses for (a) insulin clamp plasma glucose and (b) GIR are shown for CTRL (squares) and HS (triangles) groups. (c) Ra, (d) Rd, (e) HGU and (f) R′g are shown following saline (white bars) or insulin (black bars) infusion. Data are means ± SEM for n = 9–11 per group. *p < 0.05 vs respective saline. † p < 0.05 vs CTRL-insulin
Insulin-mediated Rd (Fig. 2d) was 20% lower in HS rats (23.8 ± 0.8 vs 18.2 ± 0.5 mg kg−1 min−1, p < 0.001). While insulin reduced Ra in both CTRL (Fig. 2c; p < 0.001) and HS (p < 0.001) groups compared with saline, HS displayed lower Ra following both saline (p = 0.076) and insulin (p = 0.002) infusion.
Muscle glucose uptake
Insulin increased hindlimb glucose uptake (HGU; Fig. 2e) in both CTRL (p < 0.001) and HS (p < 0.001) groups compared with respective saline groups. While no difference in HGU was detected following saline between the groups (p = 0.268), HS rats exhibited reduced insulin-stimulated HGU (0.62 ± 0.08 vs 0.48 ± 0.02 μmol/min; p = 0.028).
Consistent with HGU, insulin increased R′g (Fig. 2f) in both CTRL (p < 0.001) and HS (p < 0.001) groups compared with saline. No difference was apparent between the groups following saline infusion (p = 0.799). However, as with HGU, insulin-stimulated R′g was 30% lower in HS rats (24.7 ± 1.1 vs 17.6 ± 1.0 μg g−1 min−1, p < 0.001).
Akt phosphorylation
Compared with saline, insulin increased hepatic Ser473-Akt phosphorylation in both CTRL (Fig. 3a; p = 0.021) and HS (p < 0.001) groups. However, the extent of hepatic Ser473-Akt phosphorylation was significantly greater in HS rats following insulin infusion (1.00 ± 0.10 vs 1.52 ± 0.16; p = 0.002).

Ser473-Akt phosphorylation in vivo. Ser473-Akt phosphorylation following saline (white bars) or insulin (black bars) infusion in (a) liver (b) or skeletal muscle for CTRL and HS groups. Data are means ± SEM for n = 6 per group. *p < 0.05 vs respective saline. † p < 0.05 vs CTRL-insulin
Similarly, insulin increased Ser473-Akt phosphorylation in the skeletal muscle of CTRL (Fig. 3b; p < 0.001) and HS (p < 0.001) groups compared with the respective saline controls. However, compared with CTRL, HS rats displayed greater stimulation of skeletal muscle Ser473-Akt phosphorylation in response to insulin (0.86 ± 0.08 vs 1.23 ± 0.16; p = 0.005).
Haemodynamic effects
Baseline FBF was significantly (p < 0.001) lower in HS compared with CTRL rats (Fig. 4a; 0.99 ± 0.04 vs 0.54 ± 0.02 ml/min). Saline infusion did not alter FBF in either CTRL (p = 0.577) or HS (p = 0.638) groups. By contrast, insulin increased FBF in both CTRL (p < 0.001) and HS (p < 0.001) groups. Despite FBF remaining lower in HS rats following insulin infusion (1.33 ± 0.10 vs 0.71 ± 0.03 ml/min, p < 0.001), the insulin-stimulated change in FBF relative to baseline was similar among the groups (30.1 ± 6.7 vs 36.9 ± 11.5%; p = 0.234).

Haemodynamic effects in vivo. (a) Baseline (diagonal lines), post-saline (white bars) and post-insulin (black bars) FBF is shown for CTRL and HS groups. (b) Microvascular recruitment in muscle is shown following saline (white bars) or insulin (black bars) infusion. Data are means ± SEM for n = 9–11 per group. *p < 0.01 vs respective saline. † p < 0.001 vs respective CTRL group
Assessment of 1-MX metabolism (a marker of microvascular recruitment) following saline revealed no difference between CTRL and HS rats (Fig. 4b; p = 0.849). In CTRL rats insulin increased 1-MX metabolism by 33% above saline (p < 0.001). By contrast, 1-MX metabolism in HS rats was unaffected by insulin infusion and was similar to the level observed following saline infusion (p = 0.750). Consequently, the extent of insulin-mediated 1-MX metabolism in HS was markedly lower compared with CTRL rats (5.36 ± 0.36 vs 3.73 ± 0.29 nmol/min; p < 0.001).
Arterial concentrations of 1-MX were higher in the HS group compared with CTRL rats (28.1 ± 1.3 vs 35.5 ± 2.1 μmol/l; p = 0.007). No discernible difference in the circulating arterial concentration of oxypurinol was detected between the groups (6.27 ± 0.32 vs 5.71 ± 0.16 μmol/l; p = 0.266). Xanthine oxidase activity in muscle homogenates was similar between CTRL and HS groups (53.8 ± 11.8 vs 61.5 ± 6.9 pmol mg−1 min−1; n = 5 per group; p = 0.501). Thus, it is likely that the moderate increase in arterial 1-MX concentration in the HS group was due to reduced kidney clearance of 1-MX rather than reduced metabolism across the hindlimb by xanthine oxidase.
Study 2: isolated constant-flow hindlimb perfusion
Myocyte glucose uptake
Since insulin-mediated R′g was reduced by the HS in vivo, we investigated whether this was due to defects in skeletal myocyte responsiveness to insulin. As such, the rate of myocyte-specific glucose uptake (Fig. 5) of CTRL and HS rats was assessed in isolated constant-flow perfused hindlimbs, a system where insulin is unable to mediate changes in microvascular blood flow. As expected, insulin increased R′g compared with saline in both CTRL (p < 0.001) and HS (p < 0.001) groups. Importantly, compared with CTRL, HS rats displayed equivalent insulin-stimulated R′g (22.4 ± 1.8 vs 20.7 ± 1.6 μg g−1 min−1; p = 0.326).

Muscle insulin sensitivity in isolated constant-flow hindlimb perfusions. R′g is shown following saline (white bars) or insulin (black bars) in CTRL and HS groups. Data are means ± SEM for n = 5–7 per group. *p < 0.05 vs respective saline
Study 3: euglycaemic–hyperinsulinaemic clamp in HS + Q rats
HS + Q rats exhibited a modest decrease in body weight compared with the untreated HS group, while epididymal fat mass and fasting plasma glucose, insulin, lactate, ET1, and NEFA concentrations were unaffected by quinapril (Table 2).
During insulin clamps no difference in glucose concentrations was detected between groups at any time point (Fig. 6a). No difference was detected between groups following insulin clamps either in plasma ET1 (3.14 ± 0.37 vs 3.45 ± 0.57 pmol/l; p = 0.651) or insulin (1,500 ± 90 vs 1,550 ± 120 pmol/l; p = 0.501) concentrations. The GIR was 30% higher in HS + Q rats at 120 min (Fig. 6b; p < 0.001). Insulin-stimulated Rd also increased in HS + Q rats despite a further, modest reduction in Ra (Table 2). The decreased Ra was apparent despite a modest reduction in hepatic Ser473-Akt phosphorylation in HS + Q rats (Fig. 6e; 1.52 ± 0.16 vs 1.21 ± 0.17; p = 0.215).

Effects of quinapril on insulin sensitivity in HS rats in vivo. Time course of (a) plasma glucose and (b) GIR during insulin clamps is shown in HS (triangles) and HS + Q (squares) groups. Post-insulin measures are shown for (c) HGU, (d) R′g, Ser473-Akt phosphorylation in (e) liver and (f) skeletal muscle, and (h) 1-MX metabolism. (g) FBF is shown at baseline (diagonal lines) and post-insulin (black). Data are means ± SEM. n = 11 for HS; n = 8 for HS + Q. *p < 0.05 vs respective baseline. ‡ p < 0.05 vs HS
Insulin-mediated HGU improved by 25% in HS + Q rats (Fig. 6c; 0.48 ± 0.02 vs 0.62 ± 0.06 μmol/min; p = 0.022) and this was mirrored by a similar increase in R′g (Fig. 6d; 17.6 ± 1.0 vs 21.8 ± 0.9 μg g−1 min−1; p = 0.007). Ser473-Akt phosphorylation in muscle was not different between groups (Fig. 6f; 1.23 ± 0.16 vs 1.11 ± 0.09 AU; p = 0.601).
While MAP was unaffected by quinapril, basal FBF was increased and thus FVR decreased following treatment (Table 2). Insulin increased FBF in HS + Q rats (Fig. 6g; 0.70 ± 0.09 vs 0.84 ± 0.12 ml/min; p = 0.02), and this increase was similar in magnitude to that observed in untreated rats (p = 0.138). Insulin-stimulated 1-MX metabolism was approximately 25% greater in HS + Q rats compared with untreated rats (Fig. 6h; 3.73 ± 0.29 vs 4.64 ± 0.40 nmol/min; p = 0.02).
Discussion
High-energy diets and physical inactivity are recognised as two of the leading contributors to obesity and insulin resistance. However, increased sodium intake is a common feature of the obesogenic or insulin-resistant environment. Typically, hypertension is the recognised pathological consequence of increased sodium intake. The results of the present study indicate that HS intake leads to insulin resistance, primarily due to vascular dysfunction in muscle, in the absence of either myocyte-specific or liver insulin resistance. For the first time these data demonstrate a new, causative link between sodium intake and insulin resistance, without the presence of hypertension.
HS intake increases the risk of type 2 diabetes, independently of hypertension or physical inactivity [35]. Other studies provide further evidence that HS intake leads to insulin resistance [1–4] without attendant hypertension [1, 2]. Paradoxically, this is associated with an increase in proximal insulin signalling (insulin receptor substrate-1/Akt) in both muscle and liver [2, 3]. This is in direct contrast to insulin resistance originating from high-fat/obesity in which insulin signalling leading to glucose uptake is depressed. For this reason, the majority of reports conclude that the development of HS-induced insulin resistance is mediated by impairments further downstream in the insulin signalling pathway. However, one aspect that has not been previously considered is the role of the vasculature as an explanation (or cause) of HS-induced muscle insulin resistance.
We have developed two different methods for assessing microvascular blood flow in skeletal muscle in vivo that have been extensively validated. The first technique relies on stoichiometric metabolism of exogenously infused 1-MX to 1-methylurate by endothelial xanthine oxidase [11, 12, 16, 18, 21–23, 29, 30]. The second is an adaptation of an ultrasound imaging technique (contrast-enhanced ultrasound) [12, 16, 19, 20, 36, 37]. Both techniques yield similar results and we have demonstrated both in humans [36, 37] and animals [12, 18] that insulin acts to increase microvascular blood flow in muscle (microvascular recruitment) to enhance glucose disposal by muscle. Importantly, this microvascular action is independent of the ability of insulin to increase net muscle flow [12], can occur as early as 15 min following insulin infusion [19] and is partly NO-dependent [16]. It should also be noted that insulin regulates blood flow to other tissues in the body, including the heart [38], skin [39], kidney [40] and adipose [41]; however, compared with skeletal muscle these tissues contribute significantly less to postprandial glucose disposal.
The influence of insulin-mediated microvascular recruitment on muscle glucose uptake is highlighted when this action of insulin is inhibited by acute infusion of vasoconstrictors [15], TNF-α [30] or lipid emulsion plus heparin [29] where there is a parallel reduction in glucose uptake. In turn, we have also demonstrated that this microvascular insulin action is blunted or absent in obese, insulin-resistant rats [21–23] and humans [17, 37]. While elevated sodium is a common feature of the obesogenic environment, unlike dietary fat the contribution of sodium to the development of insulin resistance has been relatively poorly characterised. In the present study we hypothesised that an early driving factor in the development of HS-induced insulin resistance may be the loss of microvascular insulin sensitivity/responsiveness.
During insulin clamps, HS rats exhibited whole body insulin resistance despite enhanced hepatic insulin sensitivity, skeletal muscle insulin resistance and impaired microvascular but not femoral artery responsiveness to insulin. These defects were present despite elevated hepatic and myocyte insulin signalling, a finding that conforms to previous investigations [2–4]. However, when we examined myocyte insulin sensitivity using the perfused hindleg technique (where glucose uptake is not dependent on vascular insulin action) we found no difference between CTRL and HS groups. Therefore, these data provide compelling evidence that the mechanism of sodium-derived insulin resistance is not due to signalling or metabolic defects in the myocyte. Rather, elevated sodium intake initially leads to microvascular dysfunction and this in turn acts as the main driver of muscle insulin resistance in vivo.
Dysregulation of AngII activity is a possible link between HS intake and development of vascular dysfunction and insulin resistance [6, 9, 42, 43]. Insulin stimulates microvascular recruitment by a process that is at least partly NO-dependent [16, 19]. It has been suggested that crosstalk between AngII and insulin signalling pathways in vascular endothelium may lead to endothelial dysfunction by reducing NO bioavailability [44]. Thus, we investigated whether the ACE inhibitor quinapril could prevent insulin resistance in the HS model. Our data demonstrate that quinapril improves whole body glucose disposal in HS rats by augmenting insulin-mediated glucose uptake by skeletal muscle in vivo. We hypothesise that HS impairs the insulin signalling pathway leading to NO production and that quinapril partly restores this defect. However, the microvessels involved in this defect are at the level of pre-capillary arterioles (10–50 μm in diameter) and are thus inaccessible to direct assessment of the insulin signalling pathway in the relevant cells. Nevertheless, given that HS rats exhibited a microvascular and not myocyte defect, and that quinapril treatment augmented the microvascular response to insulin, we conclude that quinapril prevents the development of HS-derived muscle insulin resistance by preserving microvascular insulin responsiveness.
HS intake affects a number of systems within the body that regulate sodium excretion/retention. These include but are not limited to the renin–angiotensin system (RAS) [45], the sympathetic nervous system [46] and altered levels of aldosterone [45]. These pathways may play a role in HS-induced insulin resistance; however, here we report that RAS plays a major role. Several studies report promising data that ACE inhibition improves insulin sensitivity, but no consensus exists as to how this occurs. Previous work has suggested that the resulting increase in bradykinin levels following ACE inhibition may be responsible for augmentation of insulin sensitivity [47]. However, we have shown that bradykinin does not affect microvascular recruitment in muscle [48], indicating that the improvements in microvascular sensitivity by quinapril in the present study are unlikely to be mediated by enhanced bradykinin levels. While we made no direct assessment of plasma renin, AngII or aldosterone in our animals, we performed AngII dose–response curves in our constant-flow pump-perfused hindleg preparation and found that HS animals had significantly increased vascular sensitivity to AngII, which was partially reversed by quinapril (ESM Fig. 1). This finding was apparent despite all three treatment groups having similar basal perfusion pressure (not significant), indicating that no major vascular remodelling had occurred following HS or HS + Q interventions. Collectively, these data implicate HS-induced vascular insulin resistance developing due to enhanced AngII signalling and/or AngII type 1 receptor levels, rather than circulating levels of AngII or bradykinin. Thus, we provide a novel mechanistic link for improvement in muscle insulin sensitivity following ACE inhibition that involves enhancement of microvascular responsiveness to insulin.
The notion that impairment of microvascular insulin responses in skeletal muscle contributes to muscle insulin resistance is supported by recent investigations using endothelial IRS-2 knockout [49] and muscle-specific vascular endothelial growth factor knockout [50] mouse models. In both models, insulin resistance developed from a loss of insulin action within the vascular network of skeletal muscle. Additionally, we recently showed that feeding rats a fatty diet leads to muscle insulin resistance and that this is driven by a defect in the microvascular responsiveness to insulin [23]. In the present study, HS rats displayed reduced insulin-mediated muscle glucose uptake in vivo despite enhancement of insulin signalling in the myocyte. These observations, along with the data from the aforementioned studies, provide compelling evidence that it is the delivery of glucose to the myocyte that is the rate-limiting factor for insulin-mediated muscle glucose uptake. Furthermore, we hypothesise that HS impaired the insulin signalling pathway leading to NO production and that quinapril partly restored this defect thus restoring insulin-mediated glucose uptake by skeletal muscle.
In conclusion, we provide fresh insight into the mechanisms by which HS intake contributes to muscle insulin resistance. Our data indicate that sodium-induced insulin resistance has origins that are vascular, rather than myocyte- or liver-derived. Treating HS rats with quinapril augmented microvascular insulin sensitivity and restored insulin-mediated muscle glucose uptake in vivo, suggesting a role for the RAS in the development of sodium-derived microvascular insulin resistance. These results imply that reductions in dietary sodium may be clinically important not only for the management of hypertension but also for insulin resistance, and highlight the significance of the vasculature as an important therapeutic target for the prevention of muscle insulin resistance.
Abbreviations
- AngII:
-
Angiotensin II
- CTRL:
-
Control
- 2DG:
-
2-Deoxy-d-[1-14C]-glucose
- ET1:
-
Endothelin 1
- FBF:
-
Femoral artery blood flow
- FVR:
-
Femoral artery vascular resistance
- GIR:
-
Glucose infusion rate
- HGU:
-
Hindlimb glucose uptake
- HS:
-
High sodium
- HS + Q:
-
High sodium + quinapril treatment
- MAP:
-
Mean arterial blood pressure
- 1-MX:
-
1-Methyl xanthine
- NO:
-
Nitric oxide
- NOS:
-
Nitric oxide synthase
- R′g:
-
Rate of muscle glucose uptake
- Ra:
-
Rate of whole body glucose appearance
- RAS:
-
Renin–angiotensin system
- Rd:
-
Rate of whole body glucose disappearance
References
Donovan DS, Solomon CG, Seely EW, Williams GH, Simonson DC (1993) Effect of sodium intake on insulin sensitivity. Am J Physiol 264:E730–E734
Ogihara T, Asano T, Ando K et al (2001) Insulin resistance with enhanced insulin signaling in high-salt diet-fed rats. Diabetes 50:573–583
Qin B, Oshida Y, Li P, Kubota M, Nagasaki M, Sato Y (2007) Voluntary running improves in vivo insulin resistance in high-salt diet-fed rats. Exp Biol Med 232:1330–1337
Ogihara T, Asano T, Ando K et al (2002) High-salt diet enhances insulin signaling and induces insulin resistance in Dahl salt-sensitive rats. Hypertension 40:83–89
Lenda DM, Sauls BA, Boegehold MA (2000) Reactive oxygen species may contribute to reduced endothelium-dependent dilation in rats fed high salt. Am J Physiol Heart Circ Physiol 279:H7–H14
Weber DS, Frisbee JC, Lombard JH (1999) Selective potentiation of angiotensin-induced constriction of skeletal muscle resistance arteries by chronic elevations in dietary salt intake. Microvasc Res 57:310–319
Ye JM, Doyle PJ, Iglesias MA, Watson DG, Cooney GJ, Kraegen EW (2001) Peroxisome proliferator-activated receptor (PPAR)-alpha activation lowers muscle lipids and improves insulin sensitivity in high fat-fed rats: comparison with PPAR-gamma activation. Diabetes 50:411–417
Pickavance LC, Brand CL, Wassermann K, Wilding JP (2005) The dual PPARalpha/gamma agonist, ragaglitazar, improves insulin sensitivity and metabolic profile equally with pioglitazone in diabetic and dietary obese ZDF rats. Br J Pharmacol 144:308–316
Zhou MS, Schulman IH, Raij L (2009) Role of angiotensin II and oxidative stress in vascular insulin resistance linked to hypertension. Am J Physiol Heart Circ Physiol 296:H833–H839
Li J, White J, Guo L et al (2009) Salt inactivates endothelial nitric oxide synthase in endothelial cells. J Nutr 139:447–451
Rattigan S, Clark MG, Barrett EJ (1997) Hemodynamic actions of insulin in rat skeletal muscle: evidence for capillary recruitment. Diabetes 46:1381–1388
Vincent MA, Dawson D, Clark AD et al (2002) Skeletal muscle microvascular recruitment by physiological hyperinsulinemia precedes increases in total blood flow. Diabetes 51:42–48
Clark MG, Wallis MG, Barrett EJ et al (2003) Blood flow and muscle metabolism: a focus on insulin action. Am J Physiol Endocrinol Metab 284:E241–E258
Baron AD, Steinberg H, Brechtel G, Johnson A (1994) Skeletal muscle blood flow independently modulates insulin-mediated glucose uptake. Am J Physiol 266:E248–E253
Rattigan S, Clark MG, Barrett EJ (1999) Acute vasoconstriction-induced insulin resistance in rat muscle in vivo. Diabetes 48:564–569
Vincent MA, Barrett EJ, Lindner JR, Clark MG, Rattigan S (2003) Inhibiting NOS blocks microvascular recruitment and blunts muscle glucose uptake in response to insulin. Am J Physiol Endocrinol Metab 285:E123–E129
Clerk LH, Vincent MA, Jahn LA, Liu Z, Lindner JR, Barrett EJ (2006) Obesity blunts insulin-mediated microvascular recruitment in human forearm muscle. Diabetes 55:1436–1442
Zhang L, Vincent MA, Richards SM et al (2004) Insulin sensitivity of muscle capillary recruitment in vivo. Diabetes 53:447–453
Vincent MA, Clerk LH, Lindner JR et al (2004) Microvascular recruitment is an early insulin effect that regulates skeletal muscle glucose uptake in vivo. Diabetes 53:1418–1423
Dawson D, Vincent MA, Barrett EJ et al (2002) Vascular recruitment in skeletal muscle during exercise and hyperinsulinemia assessed by contrast ultrasound. Am J Physiol Endocrinol Metab 282:E714–E720
St-Pierre P, Genders AJ, Keske MA, Richards SM, Rattigan S (2010) Loss of insulin-mediated microvascular perfusion in skeletal muscle is associated with the development of insulin resistance. Diabetes Obes Metab 12:798–805
Wallis MG, Wheatley CM, Rattigan S, Barrett EJ, Clark AD, Clark MG (2002) Insulin-mediated hemodynamic changes are impaired in muscle of zucker obese rats. Diabetes 51:3492–3498
Premilovac D, Bradley EA, Ng HL, Richards SM, Rattigan S, Keske MA (2013) Muscle insulin resistance resulting from impaired microvascular insulin sensitivity in Sprague Dawley rats. Cardiovasc Res 98:28–36
Bragulat E, de la Sierra A, Antonio MT, Coca A (2001) Endothelial dysfunction in salt-sensitive essential hypertension. Hypertension 37:444–448
Zhou MS, Adam AG, Jaimes EA, Raij L (2003) In salt-sensitive hypertension, increased superoxide production is linked to functional upregulation of angiotensin II. Hypertension 42:945–951
Kaplan HR, Taylor DG, Olson SC, Andrews LK (1989) Quinapril—a preclinical review of the pharmacology, pharmacokinetics, and toxicology. Angiology 40:335–350
Burnol AF, Leturque A, Girard PJ (1983) A method for quantifying insulin sensitivity in vivo in the anesthetized rat: the euglycemic insulin clamp technique coupled with isotopic measurement of glucose turnover. Reprod Nutr Dev 23:429–435
Kraegen EW, James DE, Jenkins AB, Chisholm DJ (1985) Dose-response curves for in vivo insulin sensitivity in individual tissues in rats. Am J Physiol 248:E353–E362
Clerk LH, Rattigan S, Clark MG (2002) Lipid infusion impairs physiologic insulin-mediated capillary recruitment and muscle glucose uptake in vivo. Diabetes 51:1138–1145
Youd JM, Rattigan S, Clark MG (2000) Acute impairment of insulin-mediated capillary recruitment and glucose uptake in rat skeletal muscle in vivo by TNFa. Diabetes 49:1904–1909
Rattigan S, Appleby GJ, Miller KA et al (1997) Serotonin inhibition of 1-methylxanthine metabolism parallels its vasoconstrictor activity and inhibition of oxygen uptake in perfused rat hindlimb. Acta Physiol Scand 161:161–169
Eldershaw TP, Rattigan S, Cawthorne MA, Buckingham RE, Colquhoun EQ, Clark MG (1995) Treatment with the thiazolidinedione (BRL 49653) decreases insulin resistance in obese Zucker hindlimb. Horm Metab Res 27:169–172
Ruderman NB, Houghton CR, Hems R (1971) Evaluation of the isolated perfused rat hindquarter for the study of muscle metabolism. Biochem J 124:639–651
Colquhoun EQ, Hettiarachchi M, Ye JM et al (1988) Vasopressin and angiotensin II stimulate oxygen uptake in the perfused rat hindlimb. Life Sci 43:1747–1754
Hu G, Jousilahti P, Peltonen M, Lindstrom J, Tuomilehto J (2005) Urinary sodium and potassium excretion and the risk of type 2 diabetes: a prospective study in Finland. Diabetologia 48:1477–1483
Vincent MA, Clerk LH, Lindner JR et al (2006) Mixed meal and light exercise each recruit muscle capillaries in healthy humans. Am J Physiol Endocrinol Metab 290:E1191–E1197
Keske MA, Clerk LH, Price WJ, Jahn LA, Barrett EJ (2009) Obesity blunts microvascular recruitment in human forearm muscle after a mixed meal. Diabetes Care 32:1672–1677
Sauder MA, Liu J, Jahn LA, Fowler DE, Chai W, Liu Z (2012) Candesartan acutely recruits skeletal and cardiac muscle microvasculature in healthy humans. J Clin Endocrinol Metab 97:E1208–E1212
Serne EH, Ijzerman RG, Gans RO et al (2002) Direct evidence for insulin-induced capillary recruitment in skin of healthy subjects during physiological hyperinsulinemia. Diabetes 51:1515–1522
Molinari C, Battaglia A, Bona G et al (2001) Mechanisms of the renal vasodilation caused by insulin in anesthetized pigs. Life Sci 69:1699–1708
Sjoberg KA, Rattigan S, Hiscock N, Richter EA, Kiens B (2011) A new method to study changes in microvascular blood volume in muscle and adipose tissue: real-time imaging in humans and rat. Am J Physiol Heart Circ Physiol 301:H450–H458
Zhou MS, Schulman IH, Raij L (2010) Vascular inflammation, insulin resistance, and endothelial dysfunction in salt-sensitive hypertension: role of nuclear factor kappa B activation. J Hypertens 28:527–535
Clerk LH, Vincent MA, Barrett E, Lankford MF, Lindner JR (2007) Skeletal muscle capillary responses to insulin are abnormal in late-stage diabetes and are restored by angiotensin converting enzyme inhibition. Am J Physiol Endocrinol Metab 293:E1804–E1809
Andreozzi F, Laratta E, Sciacqua A, Perticone F, Sesti G (2004) Angiotensin II impairs the insulin signaling pathway promoting production of nitric oxide by inducing phosphorylation of insulin receptor substrate-1 on Ser312 and Ser616 in human umbilical vein endothelial cells. Circ Res 94:1211–1218
Graudal NA, Hubeck-Graudal T, Jurgens G (2011) Effects of low sodium diet versus high sodium diet on blood pressure, renin, aldosterone, catecholamines, cholesterol, and triglyceride. Cochrane Database Syst Rev (11) Art.No.: CD004022. doi:10.1002/14651858.CD004022.pub3
Osborn JW, Fink GD (2009) Region-specific changes in sympathetic nerve activity in angiotensin II-salt hypertension in the rat. Exp Physiol 95:61–68
Henriksen EJ, Jacob S, Kinnick TR, Youngblood EB, Schmit MB, Dietze GJ (1999) ACE inhibition and glucose transport in insulin resistant muscle: roles of bradykinin and nitric oxide. Am J Physiol 277:R332–R336
Mahajan H, Richards SM, Rattigan S, Clark MG (2004) Local methacholine but not bradykinin potentiates insulin-mediated glucose uptake in muscle in vivo by augmenting capillary recruitment. Diabetologia 47:2226–2234
Kubota T, Kubota N, Kumagai H et al (2011) Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab 13:294–307
Bonner JS, Lantier L, Hasenour CM, James FD, Bracy DP, Wasserman DH (2013) Muscle-specific vascular endothelial growth factor deletion induces muscle capillary rarefaction creating muscle insulin resistance. Diabetes 62:572–580
Contribution statement
DP was involved in the design, conduct/data collection, analysis and writing of the manuscript. SR, SMR and MAK were involved in the design, analysis and writing of the manuscript. MAK and DP are the guarantors of this work and as such had full access to all the data in the study, and take responsibility for the integrity of the data and the accuracy of the data analysis. All authors have approved the final version.
Funding
This work was supported by the Australian Research Council, National Health & Medical Research Council of Australia, and National Heart Foundation of Australia. SR is a Senior Research Fellow of the National Health & Medical Research Council of Australia.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
(PDF 21 kb)
Rights and permissions
About this article

Cite this article
Premilovac, D., Richards, S.M., Rattigan, S. et al. A vascular mechanism for high-sodium-induced insulin resistance in rats. Diabetologia 57, 2586–2595 (2014). https://doi.org/10.1007/s00125-014-3373-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-014-3373-y